Introduction
With a sufficient supply of growth hormone (GH), obtained via recombinant DNA technology, clinicians can treat children with growth hormone deficiency (GHD), as well as those with other non-GH deficient disorders. Short stature is defined as a height of less than -2 standard deviations (SDs) compared to the average height at the corresponding sex and age1). Idiopathic short stature (ISS) is a short stature condition in subjects with a normal birth weight, normal body proportions, normal GH secretion, and no specific cause for their short stature2,3). However, ISS subjects are a heterogeneous group of children with many unidentified causes of short stature. Children with familial short stature and constitutional delay of growth and puberty are included in the ISS category4). After the 2003 approval by the U.S. Food and Drug Administration of the use of GH for children with a height SD score (HT-SDS) of less than -2.25 and a short predicted adult height (PAH), clinical interests in GH treatment for ISS children increased5). There have been several clinical reports comparing GH treatment effects on children with ISS and those with GHD, but there are few such reports for Korean subjects, and the results presented in those reports have been inconsistent6,7,8). With those inconsistencies in mind, the authors compared the growth-promoting effects of GH in both idiopathic GHD (IGHD) and ISS subjects by retrospectively reviewing medical records.
Materials and methods
1. Subjects
We reviewed retrospectively the medical records of short statured children who were treated with GH between March 2002 and March 2016. Subjects with chromosomal abnormalities, organic brain lesions, systemic diseases, or syndromes that result in growth disorders were excluded. All included subjects had a normal birth weight. Among the 56 short statured children, conventional GH provocation tests using insulin and glucagon were performed in order to classify them as either GHD or ISS. Normal GH response at the test was defined as a peak stimulated GH level above 10 ng/mL. If the GHD subject's brain magnetic resonance images appeared normal, the diagnosis was IGHD.
2. Methods
During GH treatment, auxological and biochemical parameters including bone age (BA), insulin-like growth factor-1 (IGF-1), and insulin-like growth factor binding protein-3 (IGFBP-3) were recorded every 6 months. BA was evaluated by using the Greulich-Pyle method9), while PAH was calculated by applying the Baily-Pinneau method10). Midparental height (MPH) was the average height of the parents plus 6.5 cm in boys and minus 6.5 cm in girls. The HT-SDS was calculated as the children's height minus the average height for children of the same age and sex divided by the SD11). Both MPH and PAH were calculated as SDS compared to the sex-matched adult height SD. The values of IGF-1 and IGFBP-3 were changed into age- and sex-matched SDS (IGF-1-SDS and IGFBP-3-SDS, respectively)12). Annual and total increment ratios of BA with HT-SDS, and PAH-SDS were calculated over a period of 3 years. Initially, GH treatment was provided at a dose 0.23 mg/kg/wk for the IGHD subjects and 0.23ŌĆō0.33 mg/kg/wk for the ISS subjects. The GH dosage for the subjects with ISS was adjusted according to the subject's IGF-1 level in order not to exceed a dose greater than +2 SD.
3. Statistical analysis
Statistical analyses were performed by using IBM SPSS Statistics ver. 23.0 (IBM Corp., Armonk, NY, USA). All data are expressed as mean┬▒SD values. Mann-Whitney U-test was applied to compare differences of numerical variables between the groups at each time and chi-square test or Fisher exact test was performed to compare frequencies between groups. Wilcoxon signed rank test was applied to compare differences of the variables within the groups at each time. A P-value of 0.05 or less was considered statistically significant; however, in multiple comparisons between all times, the Bonferroni correction was applied and a P-value of 0.005 or less was considered statistically significant.
Results
1. Clinical characteristics of study population at the start of GH therapy
Among the 56 subjects included in this study, there were more males (n=40, 71.4%) than females (n=16, 28.6%). At baseline, the mean HT-SDS for all subjects was -2.45┬▒0.11, and the mean IGF-1-SDS was -0.80┬▒0.06. The peak-stimulated GH level was 20.3┬▒10.6 ng/mL and 6.6┬▒2.5 ng/mL in children with ISS and IGHD, respectively (P<0.001). Similarly, the mean IGF-I-SDS value was significantly higher in children with ISS than that in children with IGHD (-0.53┬▒0.85 vs. -1.07┬▒0.77; P=0.0018). The 2 groups were not different in chronologic age (CA), height, weight, body mass index, MPH-SDS, HT-SDS, and PAH-SDS. However, children with IGHD had significantly lower BA, BA/CA ratio (Table 1).
2. Changes in clinical characteristics during GH therapy for 3 years
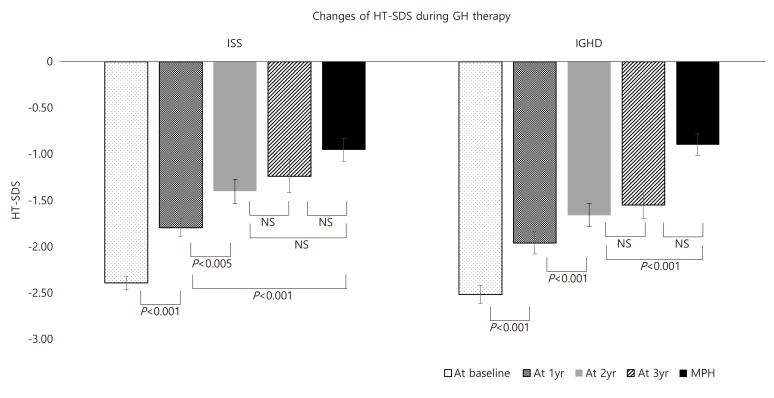
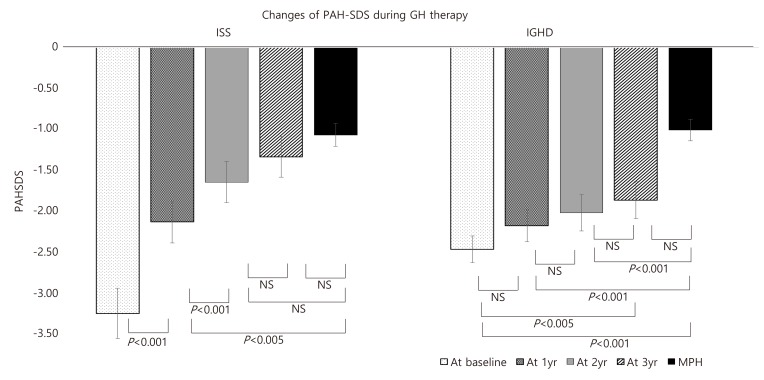
The CA, BA, BA/CA ratio, HT-SDS, PAH-SDS, IGF-1-SDS, and IGFBP-3-SDS were not significantly different between the ISS and IGHD groups after 1, 2, or 3 years of GH therapy except CA at 3 years. The GH dosage was consistently higher in the ISS group than in the IGHD group, until the end of the second year (Table 2). In both groups, the HT-SDS increased significantly until the end of second year. As a result, the increased HT-SDS was not significantly different from the MPH-SDS at the end of the second year in the ISS group, and at the end of the third year in the IGHD group (Fig. 1). The PAH-SDS increased significantly until the end of the second year in the ISS group, but there was no significant change in PAH-SDS at each year in the IGHD group. Initial PAH-SDS statistically approached MPH-SDS at the end of the second year of GH treatment in the ISS group and the third year in the IGHD group, respectively (Fig. 2).
3. Annual changes in growth parameters during GH therapy for 3 years
Annual changes in the BA/CA ratio (ΔBA/ΔCA) were significantly greater in the IGHD group than in the ISS group over the full 3-year treatment period as well as after 1, 2, and 3 years of GH therapy. However, full term and annual changes in the ΔHT-SDS/ΔCA ratio were not different between the 2 groups. Annual changes in the ΔPAH-SDS/ΔCA ratio were greater in the ISS group than in the IGHD group during the first 2 years, but not in the third year. Over the full 3-year treatment period, ΔPAH-SDS/ΔCA was significantly higher in the ISS group than in the IGHD group (Table 3).
Discussion
From our analyses, the following results were obtained; (1) before GH treatment, the IGHD group had a younger BA, lower BA/CA ratio, and lower IGF-1 level than those in the ISS group; (2) during treatment, the GH dose was consistently and significantly higher in the ISS group than in the IGHD group, but the levels of IGF-1 and IGFBP-3 in the two groups were not different; (3) although the annual BA increment was higher in the IGHD group and annual PAH-SDS increment was higher in ISS group, the annual HT-SDS increments were not different between the 2 groups; and (4) both HT-SDS and PAH-SDS in the ISS group increased significantly until the end of the second year, after which there was no significant difference between those and MPH-SDS. The HT-SDS in the IGHD group showed significant increases until the end of the second year, and it approached MPH-SDS at the end of the third year. The PAH-SDS in the IGHD group continuously increased to approach the MPH-SDS at the end of third year. With regard to PAH-SDS, the ISS group initially appeared to grow more rapidly, compared to the growth in the IGHD group, whereas, the HT-SDS results appeared to indicate that growth in both groups was similarly. However, we conclude this phenomenon is an optical illusion resulting from the BA difference. The baseline PAH-SDS was lower in the ISS group than in the IGHD group, but without statistical significance. This difference might have resulted in a significantly higher annual PAH-SDS increment in the ISS group during GH treatment, because the PAH calculation is sensitive to BA. The lower BA in the IGHD group resulted in a relatively higher PAH-SDS before treatment; thus, there was lower annual PAH-SDS increment during GH therapy.
Currently, the peak GH level used to differentiate GHD from ISS is 10 ng/mL, although this cut-off lacks physiological support13). Even though the CA, HT-SDS at diagnosis, and MPH in our ISS and IGHD groups were similar, those two groups are undeniably different, because the BA/CA ratio and the IGF-1-SDS were significantly lower in the IGHD group than in the ISS group.
When undergoing GH therapy, children with GHD usually attain a normal adult height, but children with ISS do not13,14). Lee et al.15) reported that the effect of GH on the final height of 25 children with GHD (14 idiopathic and 11 organic) was comparable to the target height. A similar result showing that the HT-SDS improved from -4.13 to +0.22 during 3.2 years of GH therapy with a dosage of 0.52ŌĆō0.62 IU/kg/wk for 35 children with GHD (13 idiopathic and 22 organic). In that study, the height at onset of puberty was associated with the improvement in final adult height16). However, according to some Korean reports, the growth-promoting effect of GH is variable in children with ISS. The final height SDS was not markedly different from the control population in a study of familial short stature with GH treatment for more than 2 years at a dose level of 0.23 mg/kg/wk6). Kang et al.7) reported that HT-SDS in ISS children improved initially with a GH dose of 0.23ŌĆō0.35 mg/kg/wk, but HT-SDS did not significantly increase after 2 years, results that were unlike those for children with GHD. However, the comparison study reported by Kim et al.8) showed similar effects on HT-SDS in children with ISS and IGHD after 1 year of therapy with the same GH dose. A clinical trial involving children with ISS demonstrated the safety and effectiveness of GH treatment, with HT-SDS increasing from -2.39 to -1.83 with daily injections 0.37 mg/kg/wk over 6-month periods17). Although we do not have final adult height data for our subjects, the HT-SDS might be expected to increase to the target range during GH treatment because HT-SDS and PAH-SDS approached the MPH-SDS at the end of the second and third treatment year.
The appropriate GH dose level in ISS is controversial and the effect of GH therapy may be dose-dependent. In 3 randomized controlled studies, that were increments of 0.51 SDS (3.7 cm) in adult height with GH treatment 0.22 mg/kg/wk for 4.4 years18), of 0.70 SDS (4.3ŌĆō5.0 cm) in adult height with dose levels of 0.23ŌĆō0.47 mg/kg/wk for 5.9 years19), and of 1.23 SDS (7.5 cm) in female adult height with a dose of 0.42 mg/kg/wk for 6.2 years20). In the present study, we used dose levels of 0.23ŌĆō0.33 mg/kg/wk, which is greater than the suggested dosage for GHD, but far below the dose levels used in previous ISS studies. The primary reason we used a relatively low dosage for the ISS children is the high cost of treatment, as the use of GH is not covered by the Korean national health insurance system. Secondarily, at the dose selected, the increased IGF-1 level in ISS group during GH therapy was thought to reflect the response of the GH-IGF-1 axis to some extent. In fact, the levels of IGF-1 and IGFBP-3 in the ISS group did increase to positive values that were similar to those in the IGHD group. For treatment effectiveness and long-term safety, a dosing regimen of GH titrated to an IGF-1 target of 0 SDS was recommended following analyses of the ╬öHT-SDS/GH dose ratio21). In another report by the same author, although the IGF-1 targets were met equally, there was significantly less height gain in ISS children than in GHD children22). This suggests that IGF-1 insensitivity or GH insensitivity may be present in ISS children. Therefore, the optimal IGF-1 target for GH treatment in non-GHD subjects needs to be elucidated.
Usually, increased effectiveness of GH therapy is correlated with a younger age at GH therapy initiation, more severe short stature, longer duration of treatment, and higher dosage23). However, the heterogeneous nature of ISS might be related to a variable growth-promoting effect. Sotos and Tokar24) organized their child subjects with ISS into familial and nonfamilial ISS groups, and reported a more favorable adult height gain in the nonfamilial ISS group. A similar result was reported in a Korean study of subjects with familial short stature5). We did not classify our ISS subjects into familial and nonfamilial ISS groups; regardless, the study results will be useful for interpreting final height gains in ISS subjects.
There are some limitations to this study. This was not a prospectively designed study comparing the effectiveness of GH treatment between children with ISS and those with IGHD. Rather, this study reported observational results for 1ŌĆō3 years of GH treatment, and we used PAH as an indicator of final adult height. Moreover, our results cannot determine whether the dose regimen used for our ISS children was superior to a higher dose recommended regardless of IGF-1 values. Park and Cohen25) proposed a treatment model that initially used a higher IGF-1 target (+2 to +3 SDS) in order to increase growth maximally, and followed that high target period by a lower IGF-1 target period for growth maintenance. However, in the present study, we did not adopt this model or the higher dose regimen because ISS includes normal short stature subjects, as well subject safety and treatment cost were of utmost importance. We did not detect any adverse effects related to the dosage we used in both groups. Another limitation of our study is that the numbers of subjects differed in each year of treatment. At the third year of treatment, half of the ISS subjects had dropped from the study, while 73% of IGHD subjects remained on GH therapy. The dropping of the subjects could bias the mean CA values, although the third year's data were obtained at almost 1 year from the second year's data acquisition time. To compensate for this type of error, the authors used SDS instead of real values.
In conclusion, both ISS and IGHD groups showed significant increases in HT-SDS and approached the genetic MPH-SDS target after 2 to 3 years of GH therapy, respectively. Initially, the lower BA/CA ratio and the lower level of IGF-1 in the IGHD group increased during the first year of therapy, after which those parameters were similar to those in ISS group. The ISS children received a slightly higher GH dose than that given to IGHD children. In the future, larger scaled, long-term studies including the final adult heights are needed to set the appropriate strategies of GH treatment for Korean ISS and GHD subjects.