Introduction
Floating-Harbor syndrome (FHS; OMIM #136140) is a rare autosomal dominant disorder characterized by short stature with facial dysmorphism, significantly delayed bone age, skeletal abnormalities, speech and language problems, and intellectual disabilities [1]. Mutations in the Snf2-related CREBBP activator protein (SRCAP) gene have been identified as causes of FHS [2]. SRCAP encodes the core catalytic component of the multiprotein chromatin remodeling SRCAP complex. Variable phenotypes of patients with FHS have been reported [3-6]. Most cases appear to be sporadic; however, autosomal dominant inheritance was reported in one familial case [7]. Although short stature is one of the main clinical manifestations, treatment with growth hormone (GH) therapy in patients with FHS is rare. Only a few reports have explored the response to GH therapy with regard to final adult height [8]. We report a 4.5-yeartreatment course with GH therapy in a Korean girl with FHS and a heterozygous c.7330C > T (p.Arg2444*) mutation in SRCAP.
Case report
A 7-year-old girl with distinct facial features and short stature visited our hospital for diagnostic evaluation. She had been born via cesarean section at 36-week gestation without perinatal problems; her birth weight was 2.7 kg (25th–50th percentile). She was the second child of nonconsanguineous, healthy parents. Her midparental height was 160 cm (father's height, 180 cm; mother's height, 153 cm). Her elder brother exhibited normal growth and psychomotor development. There was no family history of genetic diseases.
At initial presentation, the patient’s height was 105.8 cm (-3.54 standard deviation score [SDS]); her weight was 16.3 kg (-2.45 SDS), and her head circumference was 51 cm (-0.3 SDS). Her bone age was 4.5 years. She had dysmorphic facial features, which included a small, triangular face with deep-set eyes, a broad and bulbous nose, a short philtrum, and thin lips as well as hand anomalies, such as clinodactyly and a short thumb with clubbing. X-rays revealed middle phalange dysplasia and genu varum (Fig. 1).
The patient's fine motor and gross motor development were appropriate for her chronological age. She demonstrated severe intellectual disabilities as well as obsessive-compulsive and aggressive behaviors. The patient attended a special-education school and had been receiving rehabilitation treatment for the previous 2 years. Electroencephalogram, brain magnetic resonance imaging, echocardiography, and ultrasound of the kidneys were normal. Chromosome analysis indicated a 46, XX karyotype. Ophthalmological examination revealed strabismus and mild diplopia. Results of blood biochemical and urine studies were within the normal ranges. Thyroid function test results were also normal. Serum insulin-like growth factor 1 (IGF-1) and IGF-binding protein 3 levels were 223.1 ng/mL (normal range, 26–320 ng/mL) and 2481.1 ng/mL (normal range, 2,188–4,996 ng/mL), respectively. The GH peak levels provoked by insulin and L-dopa were 44.30 ng/mL and 6.07 ng/mL, respectively.
Whole-exome sequencing was performed to search for causative genes. Previous Sanger sequencing of single target genes and panel gene tests regarding the phenotypes of short stature and skeletal dysplasia presenting with intellectual disabilities revealed no pathogenic variants.
Genomic DNA was extracted from the buccal mucosa. The library was prepared to capture all exon regions of all human genes (~22,000) using the SureSelect kit (Agilent Technologies, Santa Clara, CA, USA). The captured regions were sequenced using the Illumina platform (NovaSeq, Illumina, Inc., San Diego, CA, USA). Raw genome sequencing data analysis, including alignment to the reference sequence (original GRCh37 from NCBI, February 2009), was performed with a mean depth of coverage of 100× (10×=99.2%). Variant calling, annotation, and prioritization were performed as previously described [9]. Variants with minor allele frequencies <0.05% for dominant disease association or 2% for recessive disease association were analyzed using population genome databases, such as the 1,000 genomes site (http://phase3browser.1000genomes.org), the Exome Variant Server (http://evs.gs.washington.edu/EVS/), and ExAC (http://exac.broadinstitute.org/).
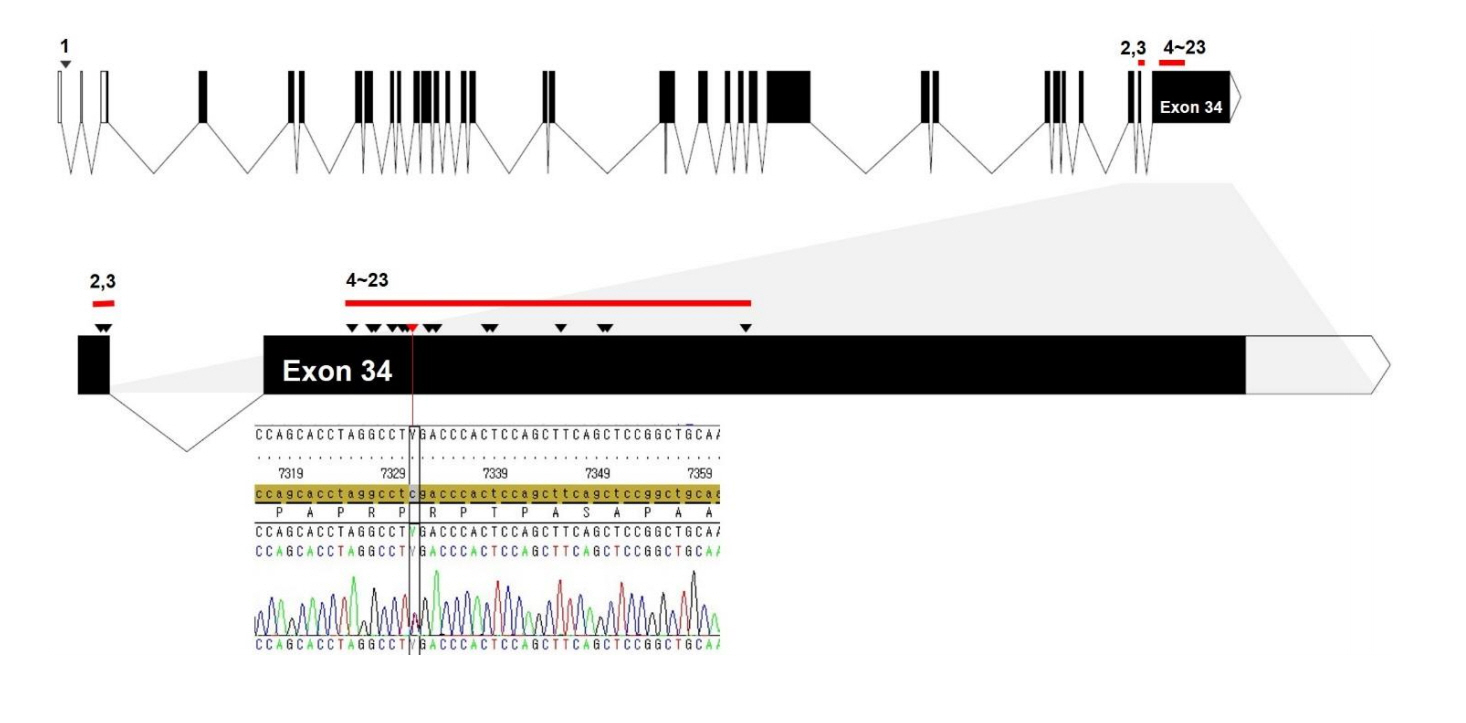
A heterozygous c.7330C > T (p.Arg2444*) mutation in SRCAP was identified and confirmed by Sanger sequencing (Fig. 2). There were no pathogenic variations in other genes related to Rubinstein-Taybi syndrome, Shprintzen-Goldberg syndrome, or other short-stature syndromes, such as those linked to mutations in the CREBBP, EP300, SKI, and ACAN genes; coverage rates greater than 20X of exons and intronic flanking regions (20 bp) were 87%, 95.6%, 93.7%, and 94.8%, respectively.
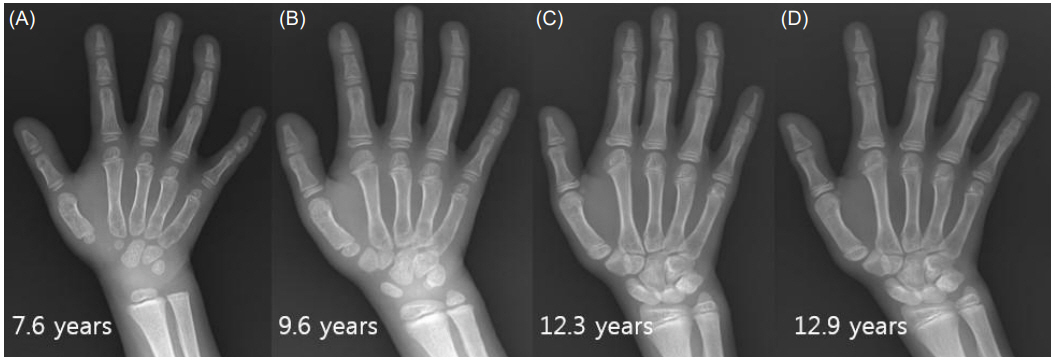
At 8 years old, the patient’s height was 112.8 cm (-3.33 SDS). Recombinant human GH replacement therapy was initiated (1.0 U/kg/wk). After 55 months of GH therapy, her height was 141.3 cm (-2.7 SDS), and she showed a modest improvement in growth velocity of 6.2 cm/yr (Fig. 3). Her bone age was consistent with her chronological age by 12 years (Fig. 4), at which point her Tanner stage was breast III. No adverse events were observed during GH treatment.
Discussion
We identified a heterozygous mutation on exon 34 of the SRCAP gene (c.7330C > T, p.R2444*) in a female Korean patient with FHS. This mutation has the highest frequency rate among FHS patients. Skeletal defects that result in altered height and posture and delayed bone age are typical of this syndrome, along with difficulties in communication and processing [1]. Other identifiable traits include dysmorphic facial features as described for our patient; however, these features can be subjective and nonspecific, complicating accurate clinical diagnosis of FHS.
SRCAP is located on chromosome 16p11.2 and includes 34 exons. SRCAP encodes an SNF2-related chromatin that remodels ATPase, which is a co-activator of CREB-binding protein (CREBBP) [10]. The CREBBP gene and its homolog, E1Abinding protein p300, play an important role in regulating cell growth and development. A mutation within CREBBP causes Rubinstein-Taybi syndrome. Therefore, the clinical features of Rubinstein-Taybi syndrome and FHS overlap. Budisteanu et al. [11] proposed a checklist of clinical features suggestive of FHS based on the main clinical features reported in the literature. Characteristic facial features and delayed language abilities are mandatory for diagnosis of FHS, while short stature, delayed bone age, skeletal anomalies, and intellectual disabilities are frequent features. Other clinical manifestations, such as behavioral problems, eye anomalies, ear anomalies, dental issues, genitourinary malformations, cardiac malformations, gastrointestinal features, seizures, and hypothyroidism, are recurrent or infrequent. Because the 2 main features of FHS, facial dysmorphism and language delay, are nonspecific, variable phenotypes exist in FHS patients. It is difficult to clinically differentiate other genetic short stature syndromes that have overlapping clinical phenotypes. Table 1 shows all the mutations associated with FHS that have been identified in the literature, including those from this study, which have been categorized according to mutation site.
Choi et al. [12] reported the first Korean case of FHS with a novel mutation (c.7732dupT, p.Ser2578Phefs*6) in SRCAP. The patient was a 7-year-old boy, and his height was 113 cm (10th–25th percentile). His language was impaired, and he had a unique facial morphology.
Short stature is the most characteristic feature of FHS; however, there are limited data on the GH–IGF-1 axis in FHS. Defects in GH secretion or activity usually do not contribute to prenatal growth inhibition, whereas IGF-1 secretion or activity is associated with prenatal growth inhibition and microcephaly. Birth size is usually normal in patients with FHS, and growth velocity begins to slow during infancy. Garcia et al. [13] suggested that FHS may lead to impaired IGF-1 signaling because of the discrepancy between the modest growth response to GH therapy and serum IGF-1 level (upper limit of the normal level during therapy). To date, over 100 FHS cases have been reported worldwide, and only a few of these patients were GH deficient [8,14]. Table 2 summarizes the results of previous studies on GH therapy in FHS patients. According to these studies, the modalities of GH therapy, age of onset, dose, treatment period, and results have been inconsistent [4,6,8,13].
In our case, the patient had normal IGF-1 level and no GH deficiency. After 55 months of GH therapy, her height SDS increased from -3.33 to -2.70 SDS, and her growth velocity was 6.2 cm/yr. Her pubertal onset and progression were not accelerated. However, her bone age progressed rapidly after puberty. In FHS, a remarkably accelerated bone age after GH therapy was previously reported [4,6]. At present, it is not known whether bone age acceleration is part of the natural course of FHS or is an adverse effect of GH therapy.
Patients with FHS manifest not only delayed bone age, but also hand abnormalities, including middle phalangeal hypoplasia, hypoplastic metacarpals, absence of a lunate, and a hypoplastic scaphoid. These findings may reflect dysregulation of chondrocyte development. Nagasaki et al. [4] suggested that SRCAP mutations can cause skeletal abnormalities due to impaired cAMP-mediated G protein-coupled receptor signaling.
In conclusion, we herein reported the clinical and molecular analysis of a 7-year-old girl with FHS and a heterozygous mutation, c.7330C>T (p.Arg2444*), in SRCAP. The patient exhibited dysmorphic facial features, severe intellectual disabilities, obsessive-compulsive and aggressive behaviors, and short stature without GH deficiency. Her height SDS was improved after 55 months of GH therapy.