Introduction
Type 1 DM is characterized by severe insulinopenia, and dependence on exogenous insulin to prevent ketosis and to preserve life1). The incidence of the disease continues to increase worldwide, and this has serious short- and long-term implications2). Glycogenic hepatopathy (GH) is a rare complication of type 1 DM, presenting as hepatomegaly and elevated serum transaminase levels in patients whose blood sugar levels are poorly controlled. In such patients, the fact that hepatomegaly may be caused by glycogen accumulation may appear to be strange, because glycogen synthesis is activated by insulin in response to high glucose levels, and insulinopenia is the principal pathophysiology of type 1 diabetes. Also, recurrent hypoglycemia is common in glycogen storage disease, and persistent hyperglycemia is a presenting symptom of diabetes.
The prevalence of liver disease in children with type 1 DM is estimated to be 4.5% and 21% using ultrasound3,4). Although there is ethnic variability in the prevalence of type 1 diabetes and these data were obtained from Egypt and Saudi Arabia, the liver disease in type 1 DM is not uncommon. Fatty liver and glycogenic hepatopathy is the predominant pathology. In the literature, GH is recognized as common in type 1 DM patients, but the underlying mechanism of development remains unclear. Until now, 7 patients with GH were reported in Korea5,6,7,8,9). All had been diagnosed with type 1 DM in childhood. Most clinician under-recognize this disease entity yet. We present a case of GH in a Korean girl with poorly controlled type 1 DM.
Case report
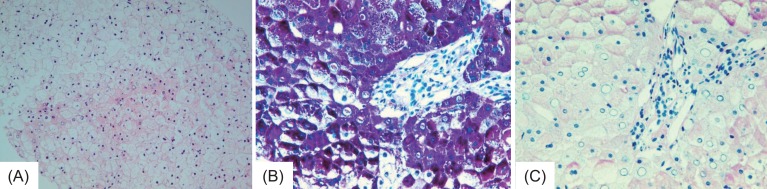
A 13-year-old female with type 1 DM was referred to our hospital complaining of abdominal pain and distention a month in duration. She had visited her local hospital and abdominal ultrasonography revealed marked hepatomegaly. She was diagnosed with type 1 DM at 11 years of age. None of her family had diabetes. Her height was 150.4 cm (10th-25th percentile) and her weight 51 kg (50th-75th percentile). Tanner stage was V (breasts and pubic hair) and bone age 15 years when she was 12 years of age (by the Greulich-Pyle method). She had been on intensive insulin therapy commencing 2 years prior (insulin aspart 8 U before meals in the morning, at noon, and in the evening; and 21 U of insulin detemir at 10 PM; total insulin dose 0.88 U/kg). However, her glycemic control was poor because she ate excessively and skipped her insulin detemir because of injection pain. She had not experienced hypoglycemia. Physical examination revealed a hard and rigid abdomen and back, and tender hepatomegaly (four fingers in breadth). Laboratory data included elevated serum liver transaminase concentrations aspartate aminotransferase, 105 U/L; alanine aminotransferase, 84 U/L and a raised lipid profile (total cholesterol, 273 mg/dL; triglycerides, 432 mg/dL; low-density lipoprotein cholesterol, 164 mg/dL). The glycated hemoglobin(HbA1C) concentration was 10.7%. Her urine contained ketones. Complete blood count data and the coagulation profile were normal. Abdominal computed tomography (CT) revealed hepatomegaly and marked liver attenuation (Fig. 1). Infectious causes were excluded, based on negative serological data for infection with cytomegalovirus, hepatitis B virus, hepatitis C virus, hepatitis A virus, and Epstein-Barr virus. The ceruloplasmin level was normal. We excluded autoimmune hepatitis. No antinuclear, antismooth muscle, anti-liver-kidney microsomal, or antimitochondrial antibodies, were detected. Liver biopsy revealed an intact lobular architecture and swollen hepatocytes with pale cytoplasm (Fig. 2). There was no evidence of inflammation, fibrosis, steatosis, or necrosis. Periodic acid-Schiff (PAS) staining revealed many PAS-positive granules within the cytoplasm of hepatocytes. Such staining after diastase digestion showed that the enzyme had destroyed the granules. The histological findings were consistent with GH. The insulin doses were adjusted to insulin aspart 8U in the morning, at noon, and in the evening, before meals; and 19 U of insulin detemir at 10 p.m. (0.84 IU/kg/day). She was re-educated on the importance of controlling nutrition and her diabetes.
With improved glycemic control, her abdominal pain and hepatomegaly decreased over 4 weeks. The HbA1C level fell to 9.5% and liver function returned to normal (aspartate aminotransferase, 44 U/L; alanine aminotransferase, 27 U/L) over 3 months.
Discussion
The most common findings in GH are hepatomegaly and elevated liver enzyme levels in blood. Our patient was obese and relatively short. She presented with hepatomegaly, elevated liver enzyme levels, and hypercholesterolemia. These symptoms differ from those of Mauriac syndrome, characterized by glycogen deposition, hepatomegaly, elevated liver enzyme levels, growth retardation, delayed puberty, cushingoid features, and hypercholesterolemia10). Our patient exhibited precocious rather than delayed puberty; she was of Tanner stage V, had menstruated at age 11 years, and her epiphyseal plate had fused 1 year prior. Our case presented with clinical and histological GH features similar to those described by others11,12,13).
GH results from excessive glycogen accumulation in the hepatocytes of patients with poorly controlled diabetes. In hepatocyte, extrahepatic glucose is in equilibrium with intrahepatic glucose due to main hepatic glucose transporter GLUT-2. Glucokinas e is the predominant glucose-phosphorylating enzyme in hepatocyte and its activity is modulated by a glucokinase regulatory protein. When blood glucose is below ~5mM, there is no significant flux through glucokinase because of its low affinity for glucose. Only when the blood sugar concentration increases above a threshold level does glucokinase start to produce glucose-6-phosphate, and it binds to liver glycogen synthase causing allosteric activation. Then protein phosphatases activate glycogen synthase. Thus hepatic glycogen synthesis is only engaged when blood glucose are high. Munns et al.13) addressed this problem, suggesting that high blood glucose concentraions are associated with infrequent doses of insulin in the patient with unstable diabetes is responsible to excess glycogen accumulation. Also, these patients take excess insulin and treat the subsequent hypoglycemic episode by administering glucose promote hepatic glycogen accumulation. This is a form of "over-glycogenosis." However, not all diabetic patients exhibiting poor glycemic control develop GH. It was suggested some regulatory protein is responsible for marked glycogen accumulation14). Tomihira et al.15) suggested that partial inhibition of liver glycogen phophorylase induced GH in patients with recurrent hepatomegaly. This is a form of "impaired glycogenolysis." The cited authors sequenced the gene encoding phosphorylase, but no defect was evident. It is however clear that fluctuations in blood sugar and insulin levels are essential elements of GH. Although the mechanism of development is unclear, GH is treated most effectively via intensive glycemic control, and the prognosis is favorable.
It is important to distinguish GH from nonalcoholic fatty liver disease (NAFLD) in clinical settings. NAFLD can progress to liver cirrhosis, whereas, with optimal glycemic control, GH does not do so, rather remitting completely12,16,17). Sweetser and Kraichely18) suggested that liver brightness on CT scans obtained without administration of contrast medium may indicate the presence of GH. Murata et al.19) used gradient-dual-echo MRI sequencing of the liver as a non-invasive tool to distinguish GH from NAFLD. However, no recognized diagnostic tool yet exists. Chatila and West16) and Brand et al.20) suggested that all diabetic patients with hepatomegaly and elevated transaminase levels should be subjected to liver biopsy. On the contrary, Munns et al.13) recommended a 4-week therapeutic trial of improved glycemic control prior to invasive investigation. In our case, liver attenuation was evident upon unenhanced CT (Fig. 1), but we performed a liver biopsy to confirm diagnosis and predict prognosis. Had we known of the features of the disease, we may not have performed a biopsy. We agree with the recommendation of Munns et al. In our case, after 3 weeks of improved glycemic control, the serum transaminase level fell to near-normal and the hepatomegaly and abdominal pain had resolved by 4 weeks. Although liver biopsy is the most accurate and reliable way by which to diagnose GH, the optimum manner of differentiating GH from NAFLD is the clinical response noted upon optimal glycemic control.
GH remains an under-recognized complication of type 1 DM. We suggest that clinicians should be aware of the disease and should monitor clinical response to optimal glycemic control. Liver biopsy may also be used to diagnose GH in patients with poorly controlled type 1 DM. Finally, it should be elucidated why some of the patients with poor controlled diabetes develop GH.