Highlights
ยท Hypoparathyroidism is a rare disorder characterized by a low corrected serum calcium or ionized calcium concentration and undetectable or inappropriately low levels of parathyroid hormone.
ยท The major goals for management are to prevent experiencing symptomatic hypocalcemia and to ameliorate the complications of the disease.
ยท Although the actions of parathyroid hormone on two major target organs: the skeleton and the kidney, neuromuscular, neurological/neuropsychiatric, renal, skeletal, cardiovascular, eye and skin complications might occur.
Introduction
Significant progress has been made in understanding the ontogenesis, functions, and diseases of the parathyroid glands. In particular, application of molecular genetic techniques has provided insight into parathyroid hormone (PTH) metabolism disorders in childhood.
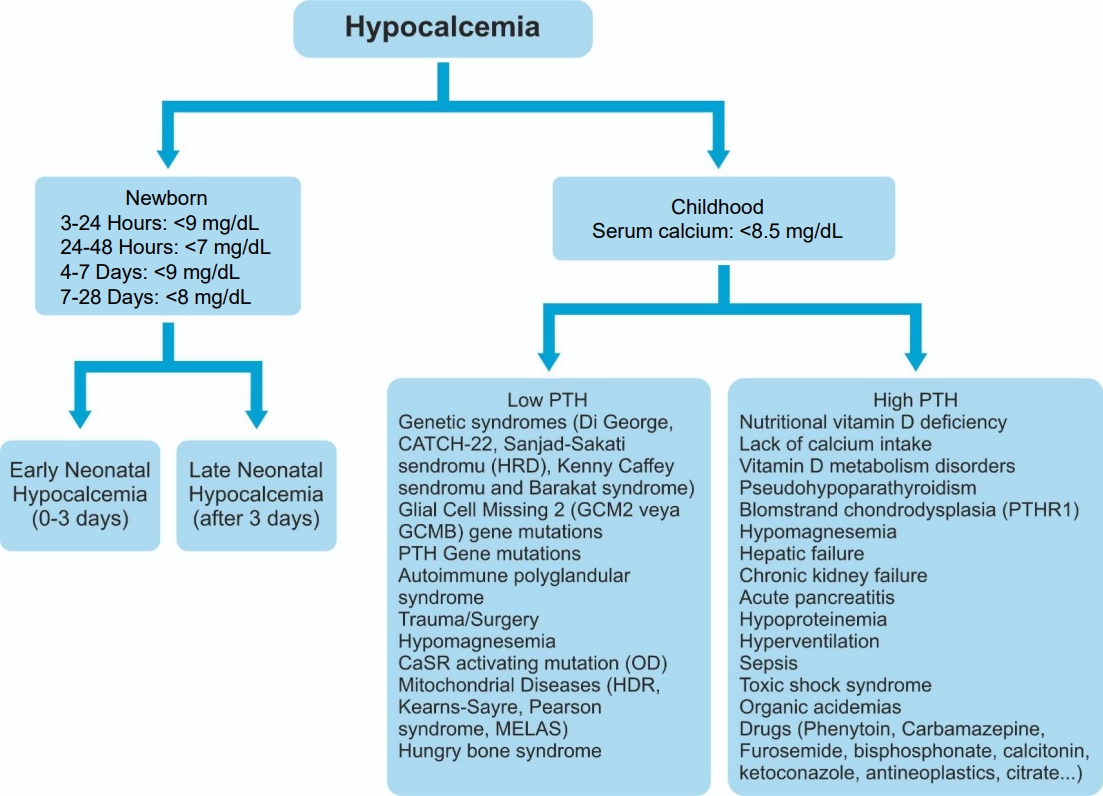
A normal or even slightly elevated PTH level may be consistent with hypoparathyroidism in the presence of hypocalcemia. In addition, serum magnesium and creatinine levels should be considered when evaluating PTH level. Severe hypomagnesemia leads to hypocalcemia [1-9]. Magnesium deficiency causes both resistance to PTH and decreased PTH secretion, resulting in hypocalcemia. In these patients, serum phosphate level is typically normal or low. Rarely, hypocalcemia develops due to decreased PTH secretion in the setting of significant hypermagnesemia. In this setting, there is also resistance to calcium and vitamin D. Serum phosphorus level is also diagnostic in the initial evaluation. Low phosphorus level suggests high PTH, and rickets should be considered in the context of high PTH and alkaline phosphatase levels [10-17]. If serum phosphorus and PTH are high, pseudohypoparathyroidism (PHP) should be considered. After confirming a hypocalcemic state, urinary calcium excretion should be evaluated. Expected urinary calcium excretion is low in hypocalcemia. If urinary calcium excretion is inappropriately normal or high, gain-of-function mutations in the autosomal dominantly inherited gene CASR should be suspected [2-9]. Diagnostic tests for hypocalcemia are shown in Table 1.
Causes of childhood hypocalcemia and associated PTH level are presented in Fig. 1 [2-9]. Genetic syndromes, parathyroid hormone gene mutations, autoimmune polyglandular syndrome, activating mutations of calcium-sensitive receptors (CaSRs), trauma or surgery-related parathyroid gland damage, and hypomagnesemia are associated with low parathyroid hormone level, whereas nutritional vitamin D and calcium deficiency, vitamin D metabolism disorders, and PHP are associated with high parathyroid hormone level [2-9]. In this review, we discuss the etiology and characteristics of hypoparathyroidism in children.
Hypoparathyroidism
Hypoparathyroidism in infancy is often associated with delayed functional development of the parathyroid gland but can be temporary and normalize within a few weeks. However, when prolonged, embryogenic defects of the parathyroid gland or disorders of PTH synthesis or secretion may be present [18-22]. Hypoparathyroidism is characterized by insufficient PTH release from the parathyroid glands to maintain serum calcium level within normal limits and unresponsiveness of target tissues despite normal serum PTH level. Hypoparathyroidism is defined as low or inappropriately normal serum PTH level. PHP is defined as unresponsiveness of target tissues despite high serum PTH level. In both cases, serum calcium level is low and phosphate level is high. Hypoparathyroidism may develop as a result of surgery or autoimmune destruction of the parathyroid glands, abnormal parathyroid gland development, disorders of parathyroid hormone production, or deficient responses to PTH. Causes of hypoparathyroidism are summarized in Table 2 [18-22]. Clinical symptoms of hypoparathyroidism are reviewed below.
Clinical symptoms of hypoparathyroidism
Patients with chronic hypoparathyroidism might remain asymptomatic due to adaptation to calcium level as low as 7.0 mg/dL [18-23]. but they may also present with various symptoms ranging from mild to severe. Mild symptoms include numbness and tingling of the extremities and perioral region, muscle cramps, and fatigue, while severe cases might present with tetany, seizure, altered mental status, cardiac rhythm disturbances, refractory congestive heart failure, bronchospasm, and laryngospasm. Acute manifestations include muscle cramps, paresthesia, numbness around the mouth, paresthesia in the extremities, tetany, calf muscle pain and stiffness, neuromuscular irritability, tetany attacks in the hands, and carpopedal spasm. Airway obstruction including laryngospasm and stridor may also be observed. Psychiatric symptoms, intellectual disability, decreased school performance, chronic fatigue, and depression may be observed in chronic hypocalcemia [18-23]. Physical examination may reveal the Chvostek sign (ipsilateral facial muscle spasm on tapping over the facial nerve), Trousseau sign (carpopedal spasm of the hand or wrist after inflation of sphygmomanometer cuff for 3 minutes), or Erb sign (increased muscle hyperexcitability) [18-24]. When hypocalcemia becomes chronic, ectodermal findings including irreversible basal ganglia calcifications, papilledema due to pseudotumor cerebri, lenticular cataracts, QT prolongation, onychoshizia, and delayed teething may be observed [18-24].
Neonatal hypoparathyroidism is typically symptomatic and presents as late neonatal hypocalcemia [18-23]. In newborns, hypocalcemia is associated with feeding difficulties, vomiting, lethargy, exaggerated startle, myoclonic beats, convulsion, apnea, tachypnea, tachycardia, cyanosis, laryngospasm, and heart failure.
1. Hypoparathyroidism after acute surgery
Acquired hypoparathyroidism develops as a result of damage to the parathyroid glands after surgery or as a result of autoimmunity [18-21]. The most common cause in adults is surgery. Postoperative hypoparathyroidism develops due to unintentional removal of the parathyroid glands or damage to their blood supply during radical neck dissection operations for thyroid, parathyroid, or head and neck cancers [22-26]. Postoperative hypoparathyroidism may be temporary or permanent. Studies have reported that transient hypoparathyroidism develops in 20% of patients undergoing total thyroidectomy after thyroid cancer, with permanent hypoparathyroidism observed in 0.8%โ3% of cases. Permanent hypoparathyroidism is more common in patients with a large goiter, large resection, or substernal goiter. Transient hypoparathyroidism is typically due to removal of one or more of the parathyroid glands or impaired blood supply during surgery [21-26].
1) Treatment of hypoparathyroidism after acute surgery
Acute hypoparathyroidism may develop after total or near-total thyroidectomy. Therefore, serum calcium and serum albumin levels should be measured on the evening prior to surgery and the next day. In patients with acute hypoparathyroidism, serum calcium and PTH levels may decrease rapidly, leading to severe symptoms [18-22]. Children with convulsions, tetany, or markedly prolonged QT on electrocardiography should be treated promptly with intravenous calcium gluconate (90-mg elemental calcium/10 mL). Intravenous doses of calcium gluconate should not exceed 2 mL/kg and should be given over 10 minutes with cardiac monitoring. The maximum dose of calcium gluconate that can be given at one time is 20 mL. If neuromuscular irritability persists, calcium gluconate administration can be repeated after 6โ8 hours, or elementary calcium infusions can be initiated at a rate of 1โ3 mg/kg/hr. Once serum calcium level normalizes, intravenous calcium supplementation should be discontinued promptly. In some centers, direct calcium and calcitriol therapy are started immediately after the operation for patients at high risk of postsurgical hypocalcemia. In a study by Asari et al. [19], a PTH level less than 15 pg/mL or a serum calcium level of 7.6 mg/dL on the second day after total thyroidectomy was associated with postoperative hypocalcemia [19].
2. Developmental disorders of the parathyroid gland
Mutations in genes involved in the development of the parathyroid glands may also lead to hypoparathyroidism. In such cases, hypoparathyroidism may present with isolated or congenital multisystem anomalies or as PTH resistance syndromes. Isolated congenital hypoparathyroidism can be X-linked, autosomal dominant, or autosomal recessive [18-22].
Parathyroid gland development (GCMB gene) defects and genetic disorders affecting PTH synthesis (PTH gene) or secretion (CaSR) may cause hypoparathyroidism [27]. The onset of hypoparathyroidism is most common in childhood but may be observed in patients aged 1 month to 30 years. Activating mutations in CaSR are the most common cause of mild isolated hypoparathyroidism and lead to reduced PTH secretion (low or low-normal PTH level) with normal serum calcium level. Due to the absence of PTH activity, renal calcium reabsorption is decreased, leading to hypercalciuria.
1) Isolated developmental disorders of the parathyroid gland
Familial isolated hypoparathyroidism (MIM#146200) includes hereditary hypoparathyroidism without accompanying complex syndromes or other organ involvement. As this condition is sporadic, it can be autosomal dominant, recessive, or X-linked recessive and develops due to dysgenesis of the parathyroid glands. To date, mutations in 3 genes, GCM2, PTH, and SOX3, have been shown to contribute to the development of isolated hypoparathyroidism [27-31].
(1) Glial Cell Missing 2 (GCM2 or GCMB) gene (MIM*603716) is located on chromosome 6p24.2, and mutations of this gene leading to loss of function are a common cause of isolated congenital hypoparathyroidism. GCM2 is a 5-exon gene encoding a DNA-binding transcription factor comprising 506 amino acids. GCM2 has also been shown to affect the expression of CASR and PTH genes. GATA-binding protein 3 (GATA3), which is mutated in Barakat (hypoparathyroidism, deafness, renal [HDR]) syndrome, is necessary for GCM2 gene expression. In humans, GCM2 homozygous inactivating or heterozygous dominant negative mutations are associated with early-onset hypoparathyroidism [27,32] and can be inherited in recessive or dominant modes.
(2) The PTH gene is located in the region of chromosome 11p15 (MIM*168450), and inactivating mutations may cause hypoparathyroidism. Mutations in the PTH gene inhibit the release of PTH by affecting the conversion of preproPTH to active PTH and translocation and exocytosis of PTH in the endoplasmic reticulum. In addition, cells may undergo apoptosis due to dysfunction of the endoplasmic reticulum. Depending on the specificity of the immunoassay used for PTH, PTH level may be detected as low, normal, or even high in patients with PTH gene mutations. Mutations in the PTH gene may show autosomal recessive or dominant inheritance [28,32-34].
(3) Variants associated with X-linked recessive earlyonset hypoparathyroidism (MIM#307700) are located on chromosomes Xq26-q27 and are likely associated with dysregulation of SOX3. Expression of SOX3 in embryonal parathyroid tissue indicates that the presence of a functional SOX3 gene is necessary for parathyroid gland development. Hypoparathyroidism due to parathyroid gland agenesis is associated with deletions or insertions in the Xq27.1 region [35].
3. Syndromic parathyroid gland development disorders
DiGeorge syndrome (DGS) was first described by DiGeorge in 1965. DGS (MIM#188400) is the most common disease associated with parathyroid gland dysgenesis in newborns and infants. DGS often develops due to microdeletions in the region of chromosome 22q11.2 in which more than 35 genes are located (del22q11.2:DGCR). 22q deletion syndrome is one of the most common microdeletion syndromes, with an incidence of 1 per 4,000โ5,000 live births. DGS is a genetic disorder characterized by hypocalcemia due to parathyroid hypoplasia, recurrent infections due to thymic hypoplasia or aplasia, congenital heart defects (particularly conotruncal anomalies), and dysmorphic facial findings. Dysplastic kidney, cervical spine instability, gastrointestinal malformations such as anal atresia and esophageal atresia, and visual impairment or ocular malformations may also accompany DGS [28-30,36-42]. Hypoparathyroidism is seen in 60% of cases of DGS. While hypocalcemia, hyperphosphatemia, and low serum PTH level are observed in the neonatal period, complications due to hypocalcemia are not observed due to compensatory hyperplasia of the parathyroid glands over time. However, patients with DGS are thought to have latent hypoparathyroidism, which can lead to hypocalcemia in stressful situations [28-30,36-41]. In addition to DGS, chromosome 22q11.2 region deletions are associated with many overlapping conditions such as velocardiofacial syndrome (VCFS,MIM #192430), conotruncal anomaly facial syndrome (CTAFS, MIM#217095), and CATCH22 (Cardiac defects, Abnormal face, Thymic hypoplasia, Cleft Palate, Hypocalcemia) syndrome. In these cases, ocular hypertelorism, short palpebral fissure, segmented nose structure, small mouth, low-set ears, micrognathia, malar hypoplasia, cleft palate, or isolated velopharyngeal insufficiency may be seen. In addition to craniofacial features, olfactory dysfunction, short stature, learning disability, and various psychological disorders may also be present. Different phenotypes of these syndromes can be observed in different members of the same family, indicating variable expression of 22q11.2 deletions [28-30]. Many cases of DGS occur de novo due to deletions on chromosome 22q11. Autosomal dominant inheritance in the same chromosome region has also been reported in association with unbalanced translocation and deletion [28-30]. The TBX1 gene, located in the center of the DGS region on chromosome 22q11.2, has an important role in the development of pharyngeal clefts and otic vesicles during early embryogenesis. When cases of CATCH22 syndrome clinical with no deletion of 22q11.2 region were examined, mutations in TBX1 causing heterozygous loss of function were found [42,43]. Therefore, haploinsufficiency in TBX1 alone may result in cardiac anomalies, abnormal facial appearance, thymic and parathyroid hypoplasia, or cleft palate causing velopharyngeal insufficiency [42]. UFD1L (ubiquitin fusion degradation-1 like, MIM*601754) is also associated with DGS and located in the 22q11.2 region. DGS cases due to deletion in UFD1L have also been reported [28-30,42,44]. Hypocalcemia has also been observed in cases with microduplication of chromosome 22q11.2. While the phenotype may be normal in these cases, multiple congenital anomalies, intellectual disability, autism, or schizophrenia may be observed. The cause of hypocalcemia in affected individuals has yet to be fully elucidated [45-47]. Mutations in the chromosome 22q11 region, known as the DGS1 region, may not always be detected in clinical cases of DGS. Chromosome 10p11-14 region deletions have also been shown to be associated with hypoparathyroidism and immune deficiency, and this gene region has been termed DGS2.
Barakat-HDR syndrome (MIM#146255) and HDR syndrome are due to defects in the GATA3 gene located in the chromosome 10p14 region and have autosomal dominant inheritance. Although GATA3 controls GCM2 gene expression, it also plays a critical role in the embryological development of the kidney, thymus, and otic vesicles, including the parathyroid gland. Hypoparathyroidism develops as a result of parathyroid gland dysgenesis. Hypocalcemia may be observed during the neonatal period up to late childhood. Renal cystic changes are also observed, which may lead to sensorineural hearing loss and chronic kidney disease. Steroid-resistant nephrotic syndrome, which can progress to chronic renal failure, has also been reported [30,48-50]. Cytogenetic abnormalities have been detected in the chromosome 10p14-10 promoter region in these patients; however, this region does not intersect with the DGS2 region. Heterozygous deletion, insertion, missense, or nonsense mutations in the GATA3 gene are also associated with HDR. Growth retardation, dysmorphic facial features, and congenital heart disease seen in cases with large terminal deletions in chromosome 10p are not seen in cases with isolated loss of function in GATA3.
Sanjad-Sakati syndrome (hypoparathyroidism, retardation, dysmorphism [HRD], MIM#241410) and Kenny Caffey syndrome type 1 (KCS1, MIM#244460) are due to biallelic mutations in the TBCE gene that encodes tubulin specific chaperone E (TBCE). Although these 2 syndromes differ phenotypically, they occur as a result of mutations in the same gene. Hypocalcemia, osteosclerosis, increased cortical thickness in long bones, loss of diploic distance in the skull with medullary stenosis, dysmorphic face, growth retardation, and small hands and feet are seen in KCS1. In HRD syndrome, intrauterine and postnatal growth restriction, retardation in developmental stages, hypoparathyroidism with onset in infancy, and a tendency to convulsions are observed. Medullary stenosis and other skeletal anomalies, microcephaly, microphthalmia, deeply located eyes, long philtrum, thin upper lip, flattened nasal root with beak nose, and large drooping earlobes may also be seen. HRD syndrome may present as hypocalcemia with low serum PTH level in infancy with a normal phosphaturic response to exogenous PTH. The cardiovascular system is typically unaffected in these patients. HRD syndrome has been described frequently in the Middle East, particularly in Saudi Arabia, where consanguineous marriage is common. The ฮฑ- and ฮฒ-tubulin subunits of the TBCE protein are required for normal embryogenesis of the parathyroid gland. Mutations in the TBCE gene lead to decreased microtubule formation and reduced formation of endosomal compartments essential for normal intracellular movement of proteins and organelles, such as the Golgi apparatus. Interestingly, the same mutation in TBCE (homozygous 12 bp deletion in exon 2, c.155_166del12) has been reported to be associated with both HRD and KCS1 in one family [28-30,51].
KCS2 (MIM #127000) has a similar phenotype to KCS1; however, ocular findings are evident and anterior fontanel closure is delayed. The inheritance of KCS2 is autosomal dominant and develops due to loss-of-function mutations in the FAM111A gene [51].
4. Hypoparathyroidism due to destruction of the parathyroid gland
1) Mitochondrial diseases
Mitochondrial diseases may be accompanied by endocrinological disorders such as diabetes and hypoparathyroidism. Hypoparathyroidism is most commonly observed in Kearns Sayre syndrome characterized by progressive external ophthalmoplegia, pigmentary retinopathy, atrioventricular heart block, cardiomyopathy, and proximal myopathy. As renal tubulopathy is also common in Kearns Sayre syndrome, hypoparathyroidism accompanied by renal tubular disorders may cause significant imbalances in calcium homeostasis. Mitochondrial encephalopathy, lactic acidosis, and stroke may also be seen in MELAS syndrome with hypoparathyroidism. Proximal myopathy and diabetes mellitus are also observed.
Although many maternally inherited mutations in the mitochondrial genome have been reported in these patients, their role has yet to be fully understood [28-30]. Mitochondrial diseases with multisystemic involvement in childhood are likely to be accompanied by hypoparathyroidism. A prevailing theory is that when heteroplasmy is present in many tissues, the threshold value for parathyroid dysfunction is exceeded and hypoparathyroidism occurs. While hypoparathyroidism may be sporadic due to large deletions in mitochondrial DNA, hypoparathyroidism can be inherited maternally due to MTTL1 mutations [30,50].
2) Autoimmune polyglandular syndrome type 1-APECED syndrome
Immune-mediated destruction of the parathyroid glands results in permanent hypoparathyroidism [18-22], which may be isolated or may appear as a part of autoimmune polyglandular syndrome. APECED (autoimmune polyendocrinopathy, candidiasis, ectodermal dystrophy type 1, MIM #240300) develops due to mutations in the autoimmune regulator 1 (AIRE) gene [18-22]. The 3 most important features of the syndrome are hypoparathyroidism, mucocutaneous candidiasis, and adrenal insufficiency. Typically, mucocutaneous candidiasis is the first sign in patients presenting before the age of 5 years. Subsequently, hypoparathyroidism develops in childhood and adolescence, and adrenal insufficiency develops later. Hypoparathyroidism is seen in more than 80% of cases with alopecia, which is present in one-third of patients. Additional diseases such as malabsorption, hypogonadism, pernicious anemia, chronic active hepatitis, thyroiditis, and insulindependent diabetes mellitus may occur in some patients. If hypoparathyroidism has developed due to CaSR-activating antibodies, the condition may resolve spontaneously. CaSRactivating antibodies have been detected both in patients with isolated acquired hypoparathyroidism and in patients with autoimmune polyglandular syndrome type 1. These antibodies reduce PTH secretion but do not cause destruction of the parathyroid glands [30-34]. Antibodies against NACHT (leucine rich repeat proteins, NALP5) have been detected in patients with OPS1 [28-30,51].
AIRE is located on chromosome 22q22.3, and APECED develops due to biallelic mutations that lead to loss of function. To date, 145 mutations have been identified in the AIRE gene; most of these are point mutations. The AIRE gene has 14 exons, encodes a protein of 545 amino acids, and is expressed in thymic medullary epithelial cells, lymph nodes, the spleen, and monocytes. Functionally, AIRE induces the expression of peripheral antigens in the thymus and promotes immune tolerance. In addition, AIRE acts as a E3-ubiquitin ligase, which is an important component of the protein modification, protein transport, and intracellular signaling machineries [28-30]. OPS1/APECED is common in Finland, and the p.Arg257* variant of the AIRE gene is responsible for 82% of cases. Candidiasis is more common in cases carrying the p.Arg257* mutation compared with cases with other mutations. While cases often present with mucocutaneous candidiasis, hypoparathyroidism is observed at older ages. Hypoparathyroidism typically occurs between the ages of 2 and 10 years, while adrenal insufficiency develops between the ages of 5 and 15 years. The development of hypercalcemia during treatment for hypoparathyroidism may indicate adrenal insufficiency. Severe and treatment-resistant hypomagnesemia can also accompany hypoparathyroidism due to APECED. In a Finnish cohort of 91 patients, mucocutaneous candidiasis was observed in 100% of cases (mostly during the first 2 years of life). The most common findings at presentation were mucocutaneous candidiasis (60%), hypoparathyroidism (32%), and adrenal insufficiency (5%). Symptoms of the disease may become evident at 2 months to 18 years of age. Clinical findings and age of onset of OPS1/APECED may vary even among siblings, with 10% of patients diagnosed with other findings of APECED including endocrinopathies such as autoimmune oophoritis (70%) causing primary ovarian failure, orchitis (30%), diabetes mellitus (30%), thyroiditis (30%), and hypophysitis (4%). Dermatological findings other than mucocutaneous candidiasis include alopecia, vitiligo, and rash with fever. Affected individuals may develop keratoconjunctivitis, pernicious anemia, hepatitis, and chronic diarrhea. Late manifestations of APECED are squamous cell carcinoma of the esophagus and mouth, asplenia, and interstitial nephritis. At least 2 of the 3 basic conditions (mucocutaneous candidiasis, hypoparathyroidism, or primary adrenal insufficiency) are required for diagnosis of APECED. Rarely, isolated chronic candidiasis, hypoparathyroidism, or primary adrenal insufficiency may be the sole manifestation of inactivating mutations in the AIRE gene [28-34].
3) Parathyroid gland damage due to substance storage
Other causes of destruction of the parathyroid gland are very rare. Irradiation or infiltrative and storage diseases of the parathyroid gland (hemochromatosis, Wilson disease, granulomatous diseases, or metastatic cancer) may cause destruction of the parathyroid glands, leading to hypoparathyroidism. In thalassemia major, parathyroid gland injury may occur as a result of iron accumulation due to transfusions. Isolated autoimmune hypoparathyroidism may also be seen. In a study of 194 patients with a diagnosis of thalassemia, hypoparathyroidism was observed in 18% of cases. The development of hypoparathyroidism was associated with the mean volume of blood administered per transfusion, the annual total volume transfused, splenomegaly, splenectomy, and chelation regimen. No relationship was observed between the development of hypoparathyroidism and serum ferritin values [52]. A further study of 713 cases of transfusion-dependent thalassemia (age range, 10โ62 years) reported a prevalence of hypoparathyroidism of 13.2% [53].
Other rare causes of acquired hypoparathyroidism are iron accumulation due to hemochromatosis, copper storage due to Wilson disease, infiltration, granulomatous diseases, and radiation (following treatment for Hodgkin/non-Hodgkin lymphoma or hyperthyroidism) [30].
Management of hypoparathyroidism in children
As a result of PTH deficiency in patients with chronic hypoparathyroidism, 1,25 (OH2) vitamin D synthesis decreases in the kidney [54-56]. Therefore, these patients should take lifelong vitamin D and calcium supplements. In children, 25โ50 mg/kg/day of elemental calcium therapy (calcium citrate, calcium carbonate, or calcium gluconate) should be divided into 3 or 4 doses a day. Maximum of 1,000โ2,000 mg of elemental calcium should be given daily [54-56]. If there is enough calcium in the diet, calcium supplementation may not be necessary. The preferred vitamin D metabolite for hypoparathyroidism is calcitriol as it has a short half-life and does not require 1ฮฑ hydroxylation in the kidney. Calcitriol should be started at 0.04โ0.08 ฮผg/kg/day for infants and 0.25 ฮผg/day for children. It can be increased in doses of 0.25 ฮผg every 2โ4 weeks to keep the serum calcium level within normal range. Maintenance dose is 0.25โ0.75 ฮผg/day for children aged 1โ5 years and 0.5โ2 ฮผg/day for children older than 5 years. Urine and serum calcium and phosphorus levels should be monitored during treatment. Serum calcium level should be checked every 3โ6 months. Kidney ultrasonography should be performed at regular intervals to detect early kidney stone formation. The most important complications of vitamin D and calcium therapy in patients with hypoparathyroidism are hypercalcemia, hypercalciuria, nephrolithiasis, nephrocalcinosis, and renal failure [54-56]. Hypercalciuria is the earliest sign of toxicity and may occur without the development of hypercalcemia. If calcium excretion in 24-hour urine analysis is 300 mg/day or more, calcium and vitamin D doses should be reduced. Thiazide diuretics should be added to the treatment regimen of some patients with hypercalciuria. In patients with PHP, PTH resistance is mainly in the proximal tubules. Therefore, calcium concentration should be maintained within the upper normal range (9โ10 mg/dL) to avoid the negative effects of high PTH on bone. Calcium and vitamin D therapy can decrease hypercalciuria and kidney stone development in patients with PHP. Recombinant human PTH administration may cause rare complications such as hypercalciuria and kidney stone development. In addition, recombinant human PTH therapy reduces calcium and vitamin D requirements as well as urinary calcium excretion. Therefore, its use is reasonable, especially in patients with nephrocalcinosis. However, it has not been approved for hypoparathyroidism treatment yet due to the lack of sufficient studies on the safety of long-term use in children [54-56].
In conclusion, persistent hypoparathyroidism is a condition that requires lifelong drug use. Side-effects or complications of the drugs should be monitored at regular intervals.