Highlights
┬Ę Coffin-Lowry Syndrome (CLS) is a rare X-linked disorder caused by mutations in RPS6KA3. We describe the clinical and molecular analysis of a five-year-old girl with CLS who presented with short stature, facial dysmorphism, tapering fingers, and developmental delays. This is the first Korean female identified with CLS and the first case of central precocious puberty (CPP) in CLS.
Introduction
Coffin-Lowry syndrome (CLS, OMIM # 303600) is an X-linked inherited disorder caused by loss-of-function mutations in the Ribosomal Protein S6 Kinase Polypeptide 3 (RPS6KA3) gene located in Xp22.2 [1]. CLS is a very rare disease, with an estimated incidence that varies from 1:40,000 to 1:100,000 [1,2]. Approximately 70%ŌĆō80% of cases are sporadic, whereas 20%ŌĆō30% have more than one additional affected family member [1,2]. Affected individuals display a wide range of phenotypes, including short stature, facial dysmorphism, tapering fingers, hearing deficits, intellectual disabilities, developmental delays, and progressive skeletal alterations [1,3].
A CLS diagnosis in males can be established using clinical manifestations, including intellectual disabilities, characteristic facial and skeletal findings, and radiographic findings [4]. However, females with CLS show various clinical features, ranging from tapering fingers with normal facial appearance and intelligence to remarkable facial dysmorphism with moderate intellectual disability [1,2]. These various clinical manifestations hinder CLS diagnosis in females compared to males. Also, establishing a CLS diagnosis in very young children can be much more difficult than in older patients because the physical characteristics are milder and not specific [2,4].
Here, we describe clinical and molecular analysis of a 5-year-old girl with CLS who presented with short stature, facial dysmorphism, tapering fingers, and developmental delays. We identified a novel, likely pathogenic, heterozygous variant, c.326_338delinsCTCGAGAC (p.Val109Alafs*10), in RPS6KA3 by exome sequencing. This is the first Korean female identified with CLS and the first case of central precocious puberty (CPP) in CLS. We also review the literature related to females with CLS.
Case report
A 5-year-old girl presenting with short stature and developmental delays visited Samsung Medical Center. This child was born at 40 weeks gestational age by vaginal delivery and had no history of perinatal problems. Her birth weight was 3,210 g (25thŌĆō50th percentile). The patient was the only child of nonconsanguineous healthy Korean parents. The father's height was 182 cm, and the motherŌĆÖs height was 168 cm. A newborn screening test for inherited metabolic disorders returned normal results. She presented with hypotonia and hyperlaxity of the joints at the age of 1 month.
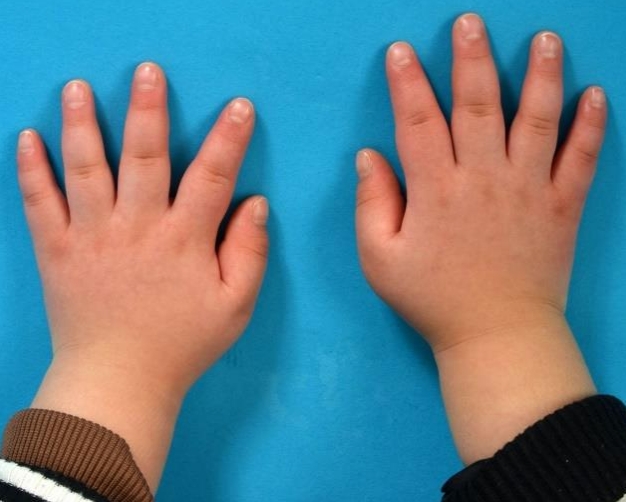
At the age of 5 years 5 months, the childŌĆÖs height was 106 cm (standard deviation score [SDS], -1.386), her weight was 20.0 kg (SDS, 0.104), and her head circumference was 50.0 cm (SDS, -0.357). SDS was estimated using the 2017 Korean National Growth Chart [5]. She showed facial dysmorphism, including a prominent forehead, hypertelorism, downward-slanting palpebral fissures, thick lips, and micrognathia. In addition, pectus excavatum and tapering stubby fingers were noted (Fig. 1).
She started to walk at 18 months of age and used 2-word sentences from the age of 5 years. At the time of her visit to our clinic, she was able to run independently and ascend stairs, but her gait was unstable, and she fell frequently. Her receptive language development was preserved, but expressive language development was delayed by about 18 months, and her pronunciation was inaccurate. The K-DST (Korean Developmental Screening Test for Infants and Children) at 65 months of age revealed global developmental delays that required further evaluation in all areas (gross motor, 4; fine motor, 3; cognition, 6; language, 8; sociality, 8; and self-control, 8).
The laboratory test results (comple te blood count, electrolytes, glucose, blood gases, hepatic and renal function tests, and urinalysis) were normal. Other endocrine evaluations were also normal: triiodothyronine, 111.62 ng/dL (60ŌĆō181 ng/dL); thyroid-stimulating hormone, 2.431 ╬╝IU/mL (0.64ŌĆō69.27 ╬╝IU/mL); free thyroxine, 1.33 ng/dL (0.89ŌĆō1.8 ng/dL); 25-hydroxyvitamin D3, 17.9 ng/mL (10ŌĆō150 ng/mL); insulin-like growth factor-1 (IGF-1), 174.5 ng/mL (70.2ŌĆō300.3 ng/mL); and alkaline phosphatase, 282 IU/L (142ŌĆō335 IU/L). Chromosome analysis revealed 46, XX. Bone age at 5 years 7 months was 2 months older than chronologic age. Lumbar scoliosis was noted on a skeletal survey.
Echocardiography and electroencephalogram (EEG) results were normal. A brain magnetic resonance imaging scan revealed multiple white matter lesions, mainly on the bilateral frontal lobe with a left paraventricular cyst. Dental examination revealed malocclusion and hypodontia (congenitally missing first molars). Ophthalmological examination showed astigmatism. The patientŌĆÖs hearing was normal.
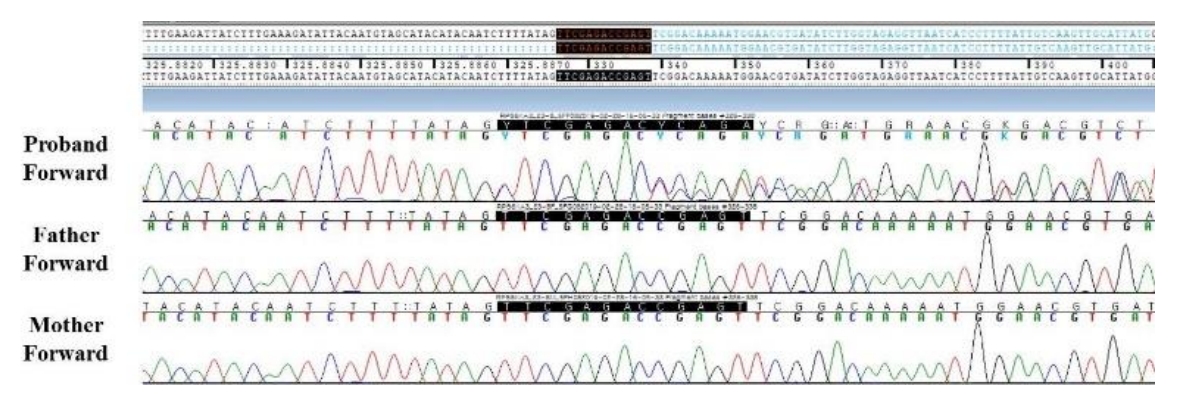
To make a diagnosis and identify underlying genetic defects, we listed the causative genes of candidate diseases based on several key symptoms (Supplementary Table 1) from the medical history and laboratory and radiographic findings. We suspected RASopathies, including Noonan syndrome and Costello syndrome. When the patient was 5 years 5 months old, exome sequencing was performed to screen for pathogenic variants of genes responsible for diseases resulting in short stature, facial dysmorphism, and developmental delays. Genomic DNA was extracted from the patientŌĆÖs peripheral blood. Approximately 57,000 target exons of a total of 4,503 genes were captured through the xGenŌōć Inherited Diseases Panel (Integrated DNA Technologies, Inc., Coralville, IA, USA) and sequenced with an Illumina NextSeq 500 (Illumina Inc., San Diego, CA, USA) for 2├Ś150 bp paired-end reads. Sequence reads were mapped to the hg19 human reference sequence using the Burrow-Wheeler Aligner version 0.7.12. The average panel depth was 166X, and 99.7% of bases were above 10X. No pathogenic variant was detected in the ATRX, HRAS, MAP2K1, PHF6, or PTPN11 gene, which are associated with RASopathies. A novel, likely pathogenic, heterozygous variant, c.326_338delinsCTCGAGAC (p.Val109Alafs*10), was identified in RPS6KA3 (Fig. 2). This variant had not been previously reported and was not found in her parents or the control databases: 1,000 genomes (http://phase3browser.1000genomes.org), Exome Variant Server (http://evs.gs.washington.edu/EVS/), and gnomAD (https://gnomad.broadinstitute.org/). The novel heterozygous variant, c.326_338delinsCTCGAGAC (p.Val109Alafs*10), is a truncated variation and is likely pathogenic according to the American College of Medical Genetics and Genomics Standards and Guidelines [6]. Eventually, the patient was diagnosed with CLS.
At the age of 7 years, the patient experienced her first sudden fall to the ground after an unexpected sound stimulus. Since that time, the frequency of fall episodes triggered by sudden sound stimuli has increased to dozens per day. Stimulus-induced drop episodes (SIDEs) were diagnosed using simultaneous surface electromyogram (sEMG) and video EEG results.
At 7 years nine months, the patientŌĆÖs breast development was Tanner stage II, and her bone age had advanced to 9 years 6 months. Her height was 122.7 cm (SDS, -0.541), and her weight was 25.0 kg (SDS, -0.237). The Bayley-Pinneau method was applied to predict an adult height of 151.8 cm, which is shorter than the midparental height of 163.5 cm. Laboratory tests yielded the following results: estradiol 1 pg/mL (<20 pg/mL), luteinizing hormone (LH) 1.1 mIU/mL (<0.02ŌĆō0.3 mIU/mL), follicle stimulating hormone 1.6 mIU/mL (0.5ŌĆō6.0 mIU/mL), and IGF-1 273.2 ng/mL (102.3ŌĆō460.1 ng/mL). The peak LH level was 5.7 mIU/mL on a gonadotropin-releasing hormone (GnRH) stimulation test. GnRH agonist therapy was initiated to treat CPP.
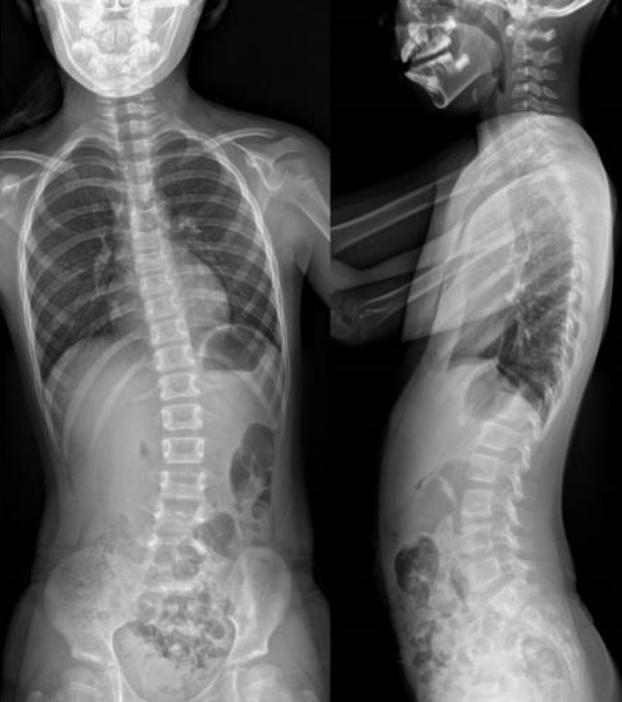
At 8 years seven months, the patient's scoliosis was 23┬░ of the Cobb angle (Fig. 3). She was able to communicate with others, write her name, and count to eight. She was able to walk independently but fell approximately 10 times a day due to hypotonia. Clobazam was prescribed to control SIDEs, but we changed that to clonazepam due to resulting drowsiness. SIDE frequency was reduced to 30% with clonazepam. Leuprolide 3.75 mg was administered once every four weeks, and pubertal development regressed.
Discussion
Descriptions of the clinical features of CLS in females are rare in the literature. Here, we presented the clinical features and natural course of a female CLS patient. CLS diagnosis in very young children is often difficult because the physical characteristics are mild and non-specific [1,4]. Moreover, female heterozygotes can show variable involvement [3,7]. CLS can be confused with other syndromes, including ATRX (alpha-thalassemia-X-linked intellectual disability) syndrome, B orjeson-Forssman-Lehmann syndrome, and Williams syndrome, which also are characterized by short stature, facial dysmorphism, and developmental delays [2,4]. Those diseases can be difficult to differentiate from CLS clinically, but a genetic evaluation can facilitate differentiation among them.
RPS6KA3 is located at Xp22.2, encompasses 22 exons, and encodes the S6K alpha 3 monomer protein that acts downstream of the mitogen-induced Ras-Activated Protein Kinase cascades. These have an important role in cell-cycle progression, differentiation, and cell survival [8]. RPS6KA3 expression activates the cAMP response element-binding protein, which plays an important role in human learning and memory. The cellular capacity to activate RPS6KA3 is key for human cognitive performance [9].
Furthermore, studies using a fibroblast cell line derived from a CLS patient indicate that the cells are defective in epidermal growth factor-induced c-fos gene expression, which regulates the differentiation and activity of specific bone cell populations [10]. The transcription factor ATF4 acts as a critical substrate of RPS6KA3, and lack of ATF4 phosphorylation can interrupt the regulatory role of ATF4 in osteoblast differentiation. Those features could explain the cognitive impairment and skeletal anomalies seen in CLS caused by RPS6KA mutations [11].
To date, more than 182 mutations have been identified in RPS6KA3 (HGMD Professional database, version 2020.4). The RPS6KA3 mutational spectrum includes 82 missense/nonsense mutations, 28 splice site variants, 33 small deletions (Ōēż20 bp), 15 small insertions (Ōēż20 bp), 1 small indel (Ōēż20 bp), 14 gross deletions, and nine gross insertions. The genotypeŌĆōphenotype correlation of CLS is unclear, and clinical features differ among patients from one family. CLS is inherited in an X-linked dominant pattern. Various findings on the skewing of X-inactivation have been demonstrated in previous studies of female CLS patients, but no associations have been found between the X-inactivation pattern and the presentation or severity of CLS manifestations [7,12]. As shown in Table 1, female heterozygotes have a wide range of observed clinical features [4,12,13]. Our patient was diagnosed with CLS at the age of 5, which is much earlier than is typical of female CLS patients (Table 1). Hyperextensible joints, tapering fingers, and hypotonia can be present at birth. These features were also observed in our patient. Early CLS diagnosis enables early detection and treatment of skeletal manifestations, such as spinal kyphosis/scoliosis. Intervention and rehabilitation in the early stages of developmental delay support the patientŌĆÖs clinical progress. Unlike males with CLS, affected females can have psychological problems. Because these symptoms progress with age, follow-up in young female patients is necessary [2,3].
SIDEs have been reported in 13%ŌĆō20% of males and 3%ŌĆō7% of females with CLS [12,14]. These episodes are characterized by a paroxysmal drop attack without loss of consciousness induced by unexpected auditory or tactile stimuli [2]. The pathophysiology of SIDEs is not well understood. Several studies have shown that epileptiform activity is absent on EEG during SIDEs. A sudden transient decrease in sEMG activity is usually observed during SIDEs, but one previous report showed increased sEMG activity [14]. We found SIDEs in hyperekplexia with increased sEMG activity in our patient.
Two previous reports of CLS have been made in Korea, 1 case in a 12-month-old male who was diagnosed clinically and 1 case in an 18-year-old male who was diagnosed by a mutation, c.889_890delAG (p.Leu298Phefs*21), in RPS6KA3 [15,16]. The patient's mother in the second case was a carrier of the same mutation, but she had no clinical signs or symptoms of CLS [16].
CLS is associated with short stature, delayed bone age, and delayed puberty [1,2]. This study differed from previous reports in that our patient showed advanced bone age, acceleration of bone maturation, and CPP. The cause of CPP in this patient is unclear and is considered spontaneous. To the best of our knowledge, no previous reports have linked CLS with CPP and treatment with a GnRH agonist, but it could be helpful in managing CLS in the future.
In conclusion, we diagnosed CLS in a young female child using next generation sequencing because diagnosis based on clinical symptoms is difficult. We report the first female Korean child with CLS and identified a novel, likely pathogenic, heterozygous variant in RPS6KA3. We suggest molecular techniques as a helpful tool in early diagnosis of rare diseases with a variable spectrum of phenotypes.