Introduction
Congenital hypothyroidism (CH) is the most common endocrine disorder in neonates and infants with an incidence of one in 2,000 to one in 4,000 newborns [1,2]. Most CH patients can be detected by newborn screening programs during the first week of life, when there may be no specific clinical symptoms or signs suggestive of CH.
Although CH can be classified as either primary or central hypothyroidism, most cases are classified as primary hypothyroidism. Primary CH can be caused by thyroid dysgenesis and thyroid dyshormonogenesis. The most common cause of CH is a developmental disorder of the thyroid gland, which occurs in 85% of all CH patients. The remaining 15% of cases are caused by defects in thyroid hormone synthesis [1].
Thyroid dyshormonogenesis can be caused by various genetic defects involving thyroid hormone biosynthesis and can present with a goiter. There are several well-known genes associated with the production of thyroid hormones, including TSHR, TG, DUOX2, DUOXA2, TPO, NIS, IYD, and pendrin genes [1]. In addition, MCT8 gene mutations, thyroid hormone resistance, maternal antithyroid antibodies, and endemic goiter can cause CH [2]. Patients with CH due to a TG gene mutation can be characterized by goitrous CH along with absent or low levels of serum thyroglobulin (Tg).
Here, we report a case of primary CH with Tg deficiency caused by a novel mutation in the TG gene.
Case report
A 15-day-old neonate was referred to our hospital with elevated thyroid stimulating hormone (TSH) (65.72 ╬╝IU/mL) found during his neonatal screening test (NST) performed at 5 days of age. He was born by Cesarean section at gestational age 38 weeks and 6 days with a birth weight of 3.4 kg. His weight, height, and head circumference were 3.9 kg (10thŌĆō25th percentile), 54.8 cm (50thŌĆō75th percentile), and 35 cm (25thŌĆō50th percentile), respectively. He had one healthy elder brother, and neither of the parents had any endocrine disorders, including thyroid disease. There was no focal neurological deficit. Physical examination revealed a wide anterior fontanelle, and there was no palpable neck mass or goiter. At that time, serum TSH was elevated to more than 100 ╬╝IU/mL (normal range, 0.5ŌĆō4.8 ╬╝IU/mL), and total T3, free T4, and Tg levels were decreased to 73.17 ng/dL (normal range, 91ŌĆō300 ng/dL), 0.228 ng/dL (normal range, 2.0ŌĆō4.9 ng/dL), and 5.53 ng/mL (normal range: cord blood 10ŌĆō115 ng/mL, infant 6ŌĆō87 ng/mL), respectively. Anti-TPO antibody (Ab) and anti-TSH receptor Ab were negative, but the level of anti-Tg Ab was mildly elevated (93.71 IU/mL; normal range, 10ŌĆō65 IU/mL). Thyroid ultrasonography revealed a normally positioned thyroid that was enlarged (right thyroid lobe: 24 mm├Ś15 mm├Ś12 mm, 2.26 mL; left thyroid lobe: 20 mm├Ś16 mm├Ś14 mm, 2.34 mL; normal range, 0.3ŌĆō1.4 mL) [3]. Blood flow was increased in the thyroid gland on color Doppler sonography. A thyroid scan was not performed.
Levothyroxine was prescribed at a dosage of 40 ╬╝g daily (10 ╬╝g/kg/day) under the diagnosis of CH since 15 days of age.
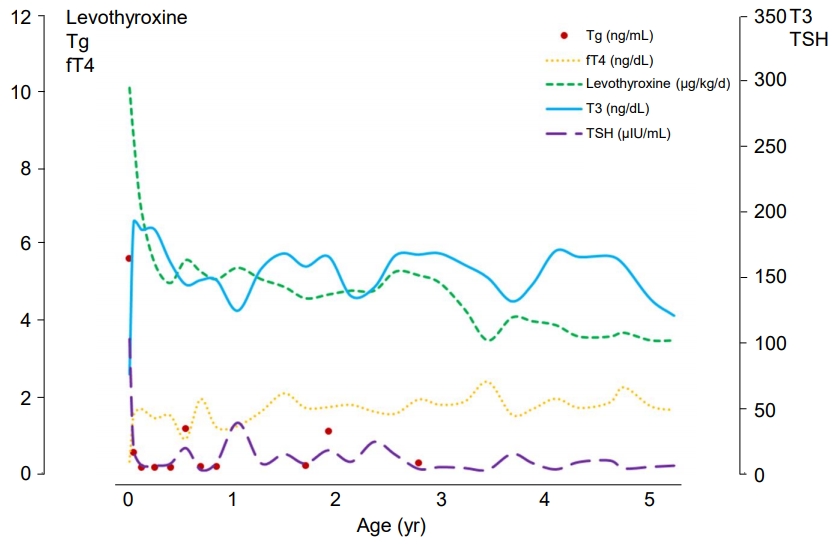
After the administration of levothyroxine for 2 weeks, serum TSH markedly decreased to 16.06 ╬╝IU/mL, and both total T3 and free T4 normalized to 189.4 ng/dL and 1.45 ng/dL, respectively. Anti-Tg Ab became negative (10 IU/mL), and his serum Tg level was 0.473 ng/mL (normal range, 10ŌĆō165 ng/mL). During follow-up examinations, he had maintained a euthyroid state with levothyroxine replacement. However, his serum Tg level was consistently low, suggesting Tg deficiency (Fig. 1).
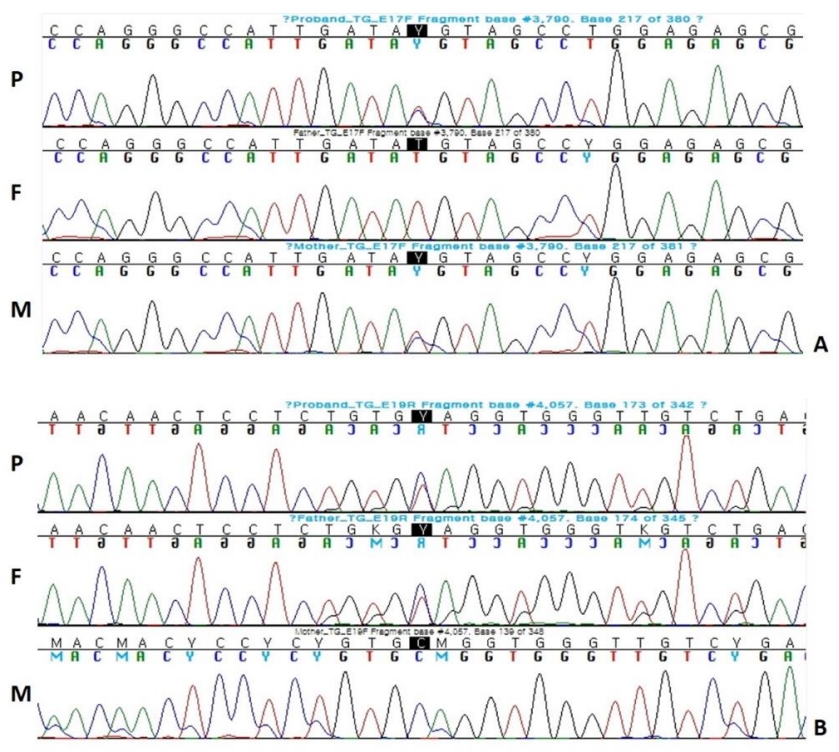
At the age of 34 months, diagnostic exome sequencing for hypothyroidism, including 23 genes (DUOX2, DUOXA2, FOXE1, GNAS, HESX1, IYD, LHX3, NKX2-1, NKX2-5, PAX8, POU1F1, PROP1, SLC16A2, SLC26A4, SLC5A5, TG, THRA, THRB, TPO, TRH, TRHR, TSHB, TSHR) associated with hypothyroidism, was performed using DNA extracted from a blood sample. A total of 238 exons from 23 genes associated with CH were analyzed, and compound heterozygous mutations were found in the TG gene. One mutation was identified in exon 17 of the TG gene and was a c.3790 T>C (p.C1264R) missense mutation. The second mutation was identified in exon 19 of the TG gene and was a c.4057C>T (P.Q1353*) nonsense mutation (Fig. 2).
Sanger sequencing of both parents' DNA samples revealed that the c.3790T>C (p.C1264R) mutation located at exon 17 was inherited from the mother, and the c.4057C>T (p.Q1353*) mutation located at exon 19 was inherited from the father, the latter of which has not been previously reported.
At the age of 3 years and 7 months, a repeat thyroid ultrasonography was performed. The thyroid was positioned normally and of normal size with the right lobe measuring 12 mm├Ś9 mm├Ś35 mm (1.97 mL) and the left lobe measuring 10.5 mm├Ś8 mm├Ś3 mm (1.36 mL) (normal range, 1.24ŌĆō2.09 mL) [4]. On color Doppler, blood flow in the thyroid gland was decreased compared to the previous thyroid sonography.
The patient is now 6 years and 8 months of age and is taking 87.5 ╬╝g of levothyroxine daily (3.1 ╬╝g/kg/day). His weight, height, and head circumference are 28.5 kg (90thŌĆō95th percentile), 127.7 cm (90thŌĆō95th percentile), and 51.8 cm (50thŌĆō75th percentile), respectively. He is experiencing normal growth and development, despite overt severe hypothyroidism detected during NST.
Discussion
This is a case report of primary CH due to Tg deficiency caused by a compound heterozygous mutation in the TG gene. We diagnosed CH based on the following findings: an increased TSH level detected during NST, low levels of T3, free T4, and Tg, and an enlarged thyroid gland without evidence of thyroid dysgenesis. The patient's presentation was clinically and biochemically compatible with CH, and the low serum Tg level was a key finding for the diagnosis of Tg deficiency. After analysis of the DNA from the patient and his parents, we confirmed that compound heterozygous mutations of the TG gene were inherited from both parents.
Tg is a large globular glycoprotein composed of 120 tyrosine units with a molecular weight of 660,000 Daltons. Tg is synthesized in the thyroid gland and plays an important role in the biosynthesis and storage of thyroid hormone. Tg is secreted through the apical surface of the thyroid follicular cells into the colloid [5]. Serum Tg levels can be elevated in neonates, Graves' disease, autoimmune thyroid disease (AITD), endemic goiter, and differentiated thyroid carcinomas, but it is markedly reduced in athyreotic infants. Patients with TG gene mutations present with low or absent levels of serum Tg, elevated TSH levels, and low levels of circulating thyroid hormones [5]. CH due to TG gene mutation appears as a heterogeneous spectrum and is inherited in an autosomal recessive manner. Therefore, patients typically have homozygous or compound heterozygous gene mutations [6]. Thyroid scintigraphy often shows an enlarged thyroid gland with high uptake [7]. If not treated appropriately, CH can result in goiter and mental retardation.
The TG gene is composed of 48 exons and is located at chromosome 8q24.2-8q24.3 [8,9]. TG gene expression is stimulated by TSH through the modulation of the intracellular level of cyclic adenosine monophosphate via the TSH receptor located at the basal membrane of the cell [5]. Generally, a TG gene mutation is in the form of homozygous or compound heterozygous because it is inherited as an autosomal recessive trait. The TG gene has multiple polymorphisms and has also been reported to play a role in the pathogenesis of AITD in the presence of certain environmental factors [10,11].
The incidence of thyroid dyshormonogenesis due to a TG gene mutation is reported to be one in 100,000 [5]. Since the first TG gene mutation was reported in 1991, more than 100 mutations of the human TG gene have been identified [6,12]. As in the present case, new mutations continue to be discovered.
In our case, the c.3790T>C (p.C1264R) mutation was located at exon 17, where cysteine was substituted by arginine. It was previously reported as a mutation affecting the intracellular transport of Tg [13]. The c.4057C>T (p.Q1353*) mutation is a novel mutation located at exon 19, where glutamine was substituted by a stop codon. Although this mutation has not been previously reported to be associated with CH, this type of mutation causes early termination in protein synthesis and has not been found in a control population (dbSNP). Therefore, this mutation can be interpreted as a pathogenic variant. In addition, this is the first case of TG deficiency diagnosed through DNA sequencing in the Republic of Korea.