Introduction
Familial hypophosphatemic rickets (FHR) is defined as a disorder resulting from a defect in renal phosphate transport, leading to phosphate wasting and hypophosphatemia [1]. FHR is a group of genetically heterogeneous disorders. It consists of X-linked dominant hypophosphatemic rickets (XLH) caused by a mutation in phosphate-regulating gene with homologies to endopeptidases on X chromosome (PHEX; MIM 300550) [2], autosomal dominant hypophosphatemic rickets caused by a mutation in FGF23 [3], autosomal recessive hypophosphatemic rickets caused by mutations in SLC34A3 [4], DMP1 [5], or ENPP1 [6], and X-linked recessive hypophosphatemic rickets caused by a mutation in CLCN5 [7]. The prevalence of XLH is approximately 1 in 20,000 [2]. PHEX is expressed in bone (particularly in osteoblasts), muscle, lung, liver, and gonads and is thought to act on substances that regulate concentrations of phosphate and 1,25-dihydroxyvitamin D3 in kidneys.
A diagnosis of XLH is based on clinical manifestation, laboratory findings, and X-ray findings. Its clinical characteristics include hypophosphatemia, deformity of the lower limb, bone pain, and growth retardation. The birth height is usually normal. However, the growth rate slows in infancy and childhood, resulting in progressive short stature. Dental problems such as tooth abscesses can also be present. The laboratory findings of XLH include low serum phosphate, hyperphosphaturia, high serum alkaline phosphatase, normal serum calcium, and low 1,25-dihydroxyvitamin D3 [1,2]. The percentage of tubular reabsorption of phosphate decreases and urinary phosphate excretion increases. The radiographic features of XLH include valgus or varus deformities of the knees. Furthermore, metaphyses of the lower limbs may be widened, frayed, or cupped. Other findings, such as calcification of the tendons, ligaments, and joint capsules and flaring of the iliac wings, may also be noted in adult patients with XLH.
A wide variety of gene defects, including nonsense mutations, missense mutations, splicing site mutations, insertions, and deletions at different positions in PHEX, have been reported [2]. To date, only 17 PHEX mutations have been reported in Korea [1,8-11], but no mosaicism has been reported. In this report, we describe a de novo mosaic mutation in PHEX in a Korean patient with hypophosphatemic rickets.
Case report
The patient was born at full term with a normal height and weight. There was no family history of consanguineous marriages or specific diseases. The patient started to walk independently at 13 months of age and showed waddling gait. At the age of 3, he was admitted to a hospital due to pneumonia. At that time, his legs began to bow noticeably and hypophosphatemia and osteopenia were found, according to the caregiver's description. He had taken oral calcium, vitamin D, and Joulie's solution since the age of 3; however, biochemical tests or X-rays had not been performed regularly.
At the age of 14, the patient's height, weight, and body mass index (BMI) were 146.4 cm (standard deviation scores [SDS], -2.53), 75.4 kg (SDS, 1.59) and 35.18 kg/m2 (SDS, 2.45), respectively. The serum calcium, phosphate, alkaline phosphatase, and creatinine were 9.4 mg/dL (8.8–10.8 mg/dL), 2.1 mg/dL (2.9–5.4 mg/dL), 694 IU/L (130–525 IU/L), and 0.5 mg/dL (0.6–0.9 mg/dL), respectively. The urine parameters were as follows: calcium/creatinine ratio, 0.1 mg/mg creatinine (0.01–0.24 mg/mg creatinine); phosphate/creatinine ratio, 0.33 mg/mg creatinine (0.22–0.86 mg/mg creatinine); and creatinine level, 0.6 g/day (0.17–1.41 g/day). The maximum tubular capacity of phosphate (TmP) per unit volume of glomerular filtration rate (TmP/GFR) calculated using the Walton and Bijvoet nomogram was 0.71 mmol/L (1.15–2.44 mmol/L).
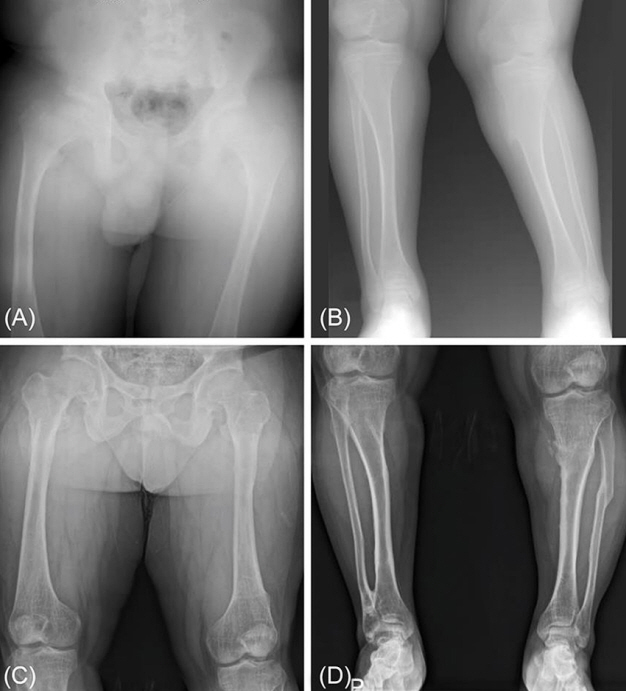
A skeletal survey showed mild coxa magna, coxa vara (Fig. 1A, B). Space of both hip joints was narrow with irregular margins; these findings suggested the early stages of osteoarthritis. He was taking 13 ng/kg/day of alfacalcidol and 2.4 mL/kg/day of Joulie's solution (73 mg/kg/day of elemental phosphorus) at that time. However, hypophosphatemia and deformities of legs were not improved during medication because his compliance for drug was very poor. He underwent hemiepiphysiodesis due to progressive valgus in both legs with short stature. The following year, he underwent corrective osteotomy again because the valgus deformity and internal torsion in his left tibia had not improved. At the age of 20, he quit all medications without consulting his physician. At the age of 26, he visited our institute to obtain a second opinion for accurate diagnosis and treatment of his disease. At that time, his height was 154.3 cm (SDS, -3.12), his body weight was 99.2 kg (SDS, 1.81), and his BMI was 41.67 kg/m2 (SDS, 2.69). Laboratory tests on serum showed the following concentrations: phosphate, 2.0 mg/dL (2.5–4.5 mg/dL); calcium, 9.4 mg/dL (8.4–10.2 mg/dL); parathyroid hormone (PTH), 94.7 pg/mL (13–104 pg/mL); 25-hydroxyvitamin D3, 15.7 ng/mL (10–150 ng/mL); alkaline phosphatase, 99 IU/L (53–128 IU/L); and creatinine, 0.81 mg/dL (0.7–1.3 mg/dL). The urine parameters were as follows: calcium/creatinine ratio, 0.1 mg/mg creatinine (≤0.22 mg/mg creatinine); phosphate/creatinine ratio, 0.1 mg/mg creatinine (0.21–0.75 mg/mg creatinine); and creatinine level, 201 mg/dL (20–370 mg/dL). The TmP/GFR of the patient was 0.80 mmol/L (1.00–1.35 mg/dL).
Radiographic findings showed mild osteoarthritis in both hip joints and multiple old fractures in both femurs, tibias, and fibulas. (Fig. 1C, D). He did not have obvious fraying and cupping shape of wrist in X-ray of both hands. Kidney ultrasonography revealed no nephrocalcinosis. Based on the medical history and laboratory/radiographic findings, FHR was strongly suspected. Because several genes (PHEX, FGF23, SLC34A3, DMP1, EPP1, and CLN5) have been known to be related to FHR, targeted gene panel sequencing for Mendelian genes was performed for molecular diagnosis.
This study was approved by the Institutional Review Board (IRB) of Samsung Medical Center (approval number: 2012-05-080-008). For molecular analysis, after obtaining informed consent from the patient, genomic DNA was extracted from peripheral blood leukocytes. Approximately 62,000 target exons of a total of 4,813 genes were captured through the Trusight One Panel (Illumina, CA, USA) and sequenced with NextSeq (Illumina) for 2×150 paired-end reads. The sequence reads were mapped to UCSC hg19 using Burrows-Wheeler Aligner–maximal exact matches (BWA-MEM, version 0.7.12). For variant calling, the Genome Analysis Tool Kit HaplotypeCaller (version 3.5) was used. Sanger sequencing was used to confirm the variant. The depth of coverage greater than 10X on the exons and intronic flanking regions (20 bp) of PHEX, FGF23, SLC34A3, DMP1, ENPP1, and CLCN5 was 99.6%, 100%, 100%, 100%, 98%, and 100%, respectively. The mean depth of the whole panel was 97X, and the depth of the mutation region was 96X.
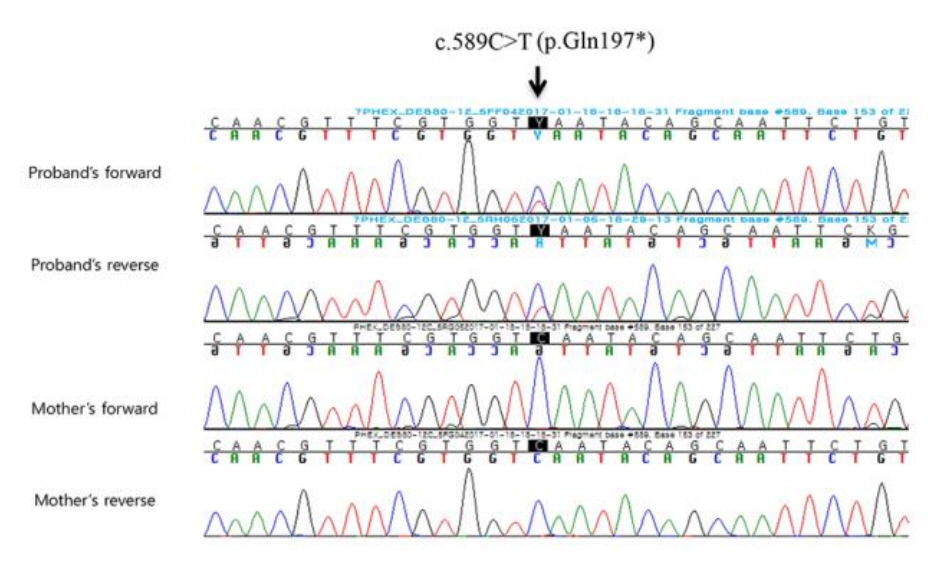
We found a novel hemizygous nonsense mutation (c.589C>T) in PHEX related to XLH that changed a glutamine residue at position 197, resulting in the formation of a stop codon; no mutations were found in the other genes. We reviewed the variant of interest for its frequency in databases, including 1,000 Genomes (http://phase3browser.1000genomes.org), Exome variant server (http://evs.gs.washington.edu/EVS/), and ExAC (http://exac.broadinstitute.org/), and minor allele frequency was 0%. The results of massive parallel sequencing led to the suspicion of mosaicism because only 38% of sequence reads showed the variant allele. Confirmatory Sanger sequencing also showed that both the wild type and the mutated base were present. No mutations in the region were found in the patient’s mother, whose phenotype was normal. Thus, this finding suggested a de novo mosaic mutation causing XLH (Fig. 2).
Discussion
FHR should be considered in the differential diagnosis of nutritional rickets, metaphyseal dysplasia, and physiologic bow legs. Laboratory findings and radiographic images are important for diagnosing FHR; however, genetic testing can be useful for an exact diagnosis.
We found a novel nonsense mutation (c.589C>T; p.Gln197Ter) in exon 5 in PHEX, which consists of 22 exons that translate into 749 amino acids. A total of 427 variants in PHEX have been reported in patients with XLH (the human gene mutation database, HGMD Professional version, access date: April 23, 2018); these include missense/nonsense mutations (37%), splice-site mutations (18%), insertion mutation (13%), and deletion mutations (8%). In Korea, 17 PHEX mutations from 20 unrelated cases have been reported as causing XLH, with the exception of the case reported here (Table 1) [1,8-11]. These comprise frameshift mutations (23.5%), nonsense mutations (23.5%), splice-site mutations (35%), and missense mutations (17.6%). All patients had skeletal abnormalities with bow legs, and 50% of them required surgical corrections. In addition, 68% of these patients had a dental disease. Table 1 shows the details of genotype, phenotype, and treatment of Korean patients with XLH carrying PHEX mutations. Several studies have reported genotype–phenotype correlations in XLH and no definite correlation has been found between disease severity and the type or location of the mutations [8,12,13].
Our patient represents the first Korean XLH case with a mosaic mutation in PHEX. Only 2 cases have reported de novo mosaic mutations in males [14,15]. One was a 4.5-year-old Chinese boy who had been suffering from gait abnormalities and bone pain since the age of 2 [14]. He was diagnosed with XLH with a de novo splice-site mutation and mosaic pattern in PHEX. A deletion of 4 bp in exon 18 led to a frameshift mutation, p.Gly590GlufsTer28, resulting in a premature stop codon. Another PHEX mosaic variant was c.1669C>T (p.Arg567Ter), which was found in a father and one of his daughters, both of whom had bow legs; another of his daughters had normal phenotype and genotype [15]. Mosaicism analysis using leukocyte and hair root DNA showed that the father had somatic and germline mosaicism of the PHEX mutation. The phenotype of mosaic mutation is considered to be milder than non-mosaicism. However, in the second mosaic mutation case, severity of clinical features and percentage of mutant allele in single-nucleotide primer extension and DHPLC analysis of leukocyte DNA of proband and her farther were similar. Therefore, other factors, including the ratio of the mutation allele, should be considered in determining the phenotype.
PHEX involves in the downregulation of FGF23. FGF23 inhibits the renal production of 1,25-dihydroxyvitamin D3 and reabsorption of phosphate by lowering 1-α-hydroxylase in the renal proximal tubule [16]. Therefore, the medical treatment of XLH includes oral phosphate supplementation and active vitamin D analogs (alfacalcidol or calcitriol) [17]. However, the aim of oral medication is not to normalize serum phosphate level, but to improve clinical symptoms, such as bone pain and short stature. If asymptomatic pediatric patients do not receive treatment, their clinical symptoms are likely to begin before the age of 1 year. In symptomatic children, growth failure and skeletal abnormalities tend to be more aggravated [11]. In asymptomatic adults, the need for medical treatment is not obvious because there has been no evidence of clinical benefits or long-term outcomes. In addition, the treatment has side effects, including nephrocalcinosis and hyperparathyroidism. Severe cases require surgery such as osteotomy or epiphysiodesis because most symptomatic patients may be partially treated with medically treatment. Thus, indications for medical treatment primarily include those in pediatric patients, symptomatic adults, and patients who need high amounts of calcium and phosphorus, such as pregnant women and perioperative orthopedic surgery patients [17]. In addition, it is necessary to monitor serum calcium, phosphate, vitamin D, and PTH and perform ultrasonography of the kidney and urinary tract while administering medications. Recently, a phase 3 clinical trial of human monoclonal antibody treatment for XLH patients was initiated in the US, the EU, Canada, Japan, and Korea (Study of KRN23 in adults with XLH; ClinicalTrials.gov Identifier NCT02526160). This study included 134 patients aged 18 to 65 years. KRN23 is an investigational recombinant fully humanized monoclonal IgG1 antibody against FGF23 [18]. In a phase 1/2 study, the subcutaneous administration of KRN23 significantly increased TmP/GFR, serum phosphate, and 1,25-dihydroxyvitamin D3. In addition, no subjects experienced an increase in nephrocalcinosis or developed hypercalciuria and hypercalcemia. However, long-term studies have not been published yet and it is necessary to expand the studies to include children.
In conclusion, we identified a novel de novo nonsense mutation in PHEX in a Korean patient with sporadic XLH. Molecular analysis revealed the mosaic pattern of the mutation. This is the first report of mosaicism of a de novo mutation in PHEX in Korea and the third such report worldwide. There have been no cases of a prenatal diagnosis of FHR by paternal sperm analysis. However, in a case of another skeletal disease, osteogenesis imperfecta, paternal sperm analysis, and chorionic villi sampling revealed recurrent autosomal dominant osteogenesis imperfecta in female fetus [19]. In our case, sperm analysis is needed to identify germline mosaicism because the mutation could be transmitted to the patient's his female offspring. In addition, tissue confirmation is required to identify somatic mosaicism.