Introduction
X-linked hypophosphatemia (XLH) is the most common form of familial hypophosphatemic rickets (FHR) and it is caused by loss-of-function mutations in the PHEX gene (phosphate regulating gene with homologies to endopeptidases on the X chromosome) with a prevalence of 1 in 20,0001,2). It is inherited as a dominant disorder and characterized biochemically by hypophosphatemia due to renal phosphate wasting with inappropriately low or normal 1,25-dihydroxyvitamin D concentrations1). Autosomal dominant hypophosphatemic rickets is known to be associated with mutations in FGF23 and autosomal recessive hypophosphatemic rickets has been reported by different groups3). Individuals with the features of FHR, but with no family history of rickets (sporadic cases) are also common, and many subsequently transmit the phenotype in an X-linked dominant manner consistent with hypophosphatemia3,4). Despite the fact that PHEX mutations cause renal phosphate wasting, PHEX is not expressed in the kidney. Instead, PHEX is predominantly expressed in bones (osteoblasts and osteocytes) and teeth (odontoblasts)2). Clinical manifestations of hypophosphatemic rickets include short stature, bone pain, dental anomalies, lower extremity deformities, and radiographic evidence of rickets in children5).
Laboratory findings of the disease include low serum phosphorus levels, normal serum calcium levels, increased activity of serum alkaline phosphatases, normal or increased parathyroid hormone (PTH) levels, and inappropriately normal or decreased levels of 1,25-dihydroxyvitamin D35,6). The PHEX gene, located on Xp22.1, consists of 22 exons that translate into a 749 amino acid protein, which is a member of the peptidase M13 family of membrane-bound metalloproteases7,8). The PHEX gene consists of a cytoplasmatic region, a transmembrane, and an extracellular domain6). Since the identification of the PHEX gene, over 351 mutations have been described in patients with XLH; those mutations include nonsense mutations, missense mutations, splicing site mutations, insertions, and deletions in both coding and noncoding regions of the gene7,9). All published mutations have been catalogued in the PHEX mutation database (http://data.mch.mcgill.ca/phexdb). Only 15 mutations of the PHEX gene in Korean patients with hypophosphatemic rickets have been reported so far3,6,10). Here, we report a sporadic case of hypophosphatemic rickets for which genetic analysis has revealed a novel mutation in PHEX, and we present a review of the literature.
Case report
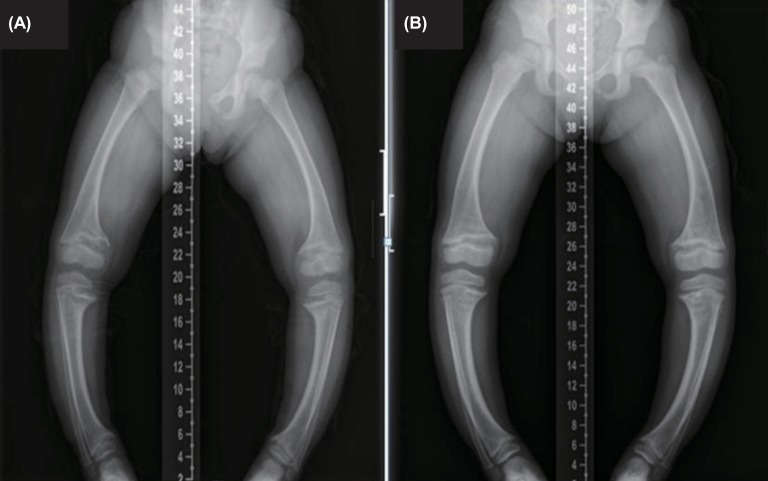
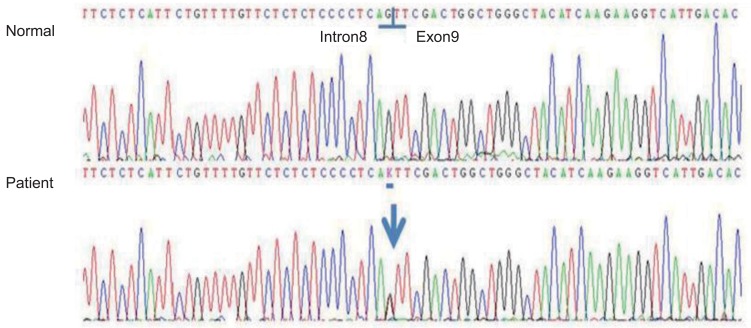
The proband was a 2-year-9-month-old Korean female who was referred to our pediatric endocrinology clinic for bilateral bowleg. She had been born at term to a 30-year-old woman whose pregnancy was uneventful. The proband's birth weight was 3,850 g and her body length was 49 cm. She was born of a nonconsanguineous marriage at full term. On admission, her height was 85.3 cm (3th-5th percentile) and her body weight was 13.5 kg (50th-75th percentile). Physical examination revealed genua vara and cafe au lait spots on the abdomen. Dental defects, including formation of abscesses and recurrent extractions, were shown in the patient. Her vision and hearing were normal. Laboratory analyses of serum revealed the following values (normal range): inorganic phosphate, 2.5 mg/dL (3.8-6.5 mg/dL); calcium, 9.4 mg/dL (8.8-10.8 mg/dL); intact-PTH, 46.53 pg/mL (9-65 pg/mL); alkaline phosphatase, 1,634 IU/L (129-291 IU/L); 25-hydroxyvitamin D3, 27.30 ng/mL (9.7-52.8 ng/mL); and 1,25-dihydroxyvitamin D3, 38.0 pg/mL (19.6-54.3 pg/mL). Radiologic findings showed widening of the proximal tibial metaphysis with medial bowing (Fig. 1). A low serum phosphorus level in combination with elevated serum alkaline phosphatase activity and normal serum calcium is suggestive of hypophosphatemic rickets and molecular analysis was done. Informed consent was obtained and genomic DNA was extracted from the peripheral blood leukocytes using a QuickGene Blood kit (Fujifilm, Tokyo, Japan). PHEX gene analysis revealed a splicing mutation, c.934-1G>T (IVS8-1G> T), which was a novel mutation (Fig. 2). However, the proband's mother had no PHEX gene mutation. All members of the proband's family were healthy.
She was treated with calcitriol 0.5 ┬Ąg/day and elemental phosphorus 2 g/day in four divided dosages. The alkaline phosphatase levels were nearly normalized (492 IU/L) within two years. At the 22-month follow-up, she was 4 years and 7 months old. At that time, her height was 98.6 cm (3th-5th percentile) and her body weight was 17.8 kg (50th-75th percentile). Complications associated with medications, such as hypercalciuria, hypercalcemia, nephrocalcinosis, renal glycosuria, and hyperparathyroidism, were not detected. Though she had been treated with medications for two years, the bowing in her legs had not improved. Therefore, the patient recently underwent surgical correction for genua vara.
Discussion
In the present study, we report a splice acceptor site mutation in the PHEX gene, c.934-1G>T (IVS8-1G>T), at the intron8 and exon9 junction. To the best of our knowledge, this mutation is novel and has not been reported. The described mutation was located in the extracellular domain in the PHEX gene, predicting that it would cause exon9 skipping. In our patient, this mutation at the exon-intron border caused subsequently severe clinical symptoms, including severe bowing of the legs, short stature, and dental defects, such as formation of abscesses and recurrent extraction. This suggests that the exon-intron border is significant in the formation of functional PHEX proteins and the mutation in this region probably contributes to the severe phenotypes. Thus, although a functional study is necessary, this mutation likely impairs production of the PHEX protein. The functional consequences of novel splice variants were predicted with the Berkeley Drosophila Genome Project (http://www.fruitfly.org/). Eighty-four de novo mutations reported in the PHEX gene in patients with sporadic hypophosphatemic rickets are summarized in Table 1. Among the 84 patients reported with sporadic hypophosphatemic rickets, including our case, splice site mutation was frequently observed (17/84, 20%). Among the 13 patients reported with sporadic hypophosphatemic rickets in Korea, including our case, splice site mutation was most frequently observed (4/13, 30.8%). Nonsense mutations (3/13, 23.1%) and missense mutations (3/13, 23.1%) were found with the same frequencies. A large number of mutations occur randomly across mainly the extracellular domain, which is encoded by exons2-22, but a mutation in the 3'-untranslated region has also recently been identified2). Moreover, the frequency of de novo mutations has been estimated to be approximately 20%11,12,13). These findings suggest that the PHEX gene appears to be particularly prone to mutations for unknown reasons13). In the present study, the patient was female and the de novo mutation was found to be a sporadic case. The female dominance in sporadic cases is also observed in other ethnic backgrounds, including Asians, Europeans, and Americans7). Given that most sporadic patients are female, it is likely that mutated PHEX alleles resulted from mutagenesis in the X chromosome of the paternal germ cells, and PHEX mutagenesis in paternal germ cells is a frequent event in sporadic patients7). Therefore, further study is needed to prove this inference at the protein level with a biopsy to obtain cells for culture.
The phenotype was assessed as severity of skeletal disease (mild vs. moderate-to-severe) and severity of dental disease (mild vs. moderate-to-severe) using the following objective criteria: history of osteotomy, degree of bowing, and history of dental abscesses4). In our patient, the truncating mutation predicts a severe clinical occurrence. The girl underwent osteotomy, dental abscess was evident, and there was severe bowing of the legs, thus meeting all the major criteria set forth by Holm et al.4) to be classified as a severe skeletal phenotype. No significant correlation was found between the severity of the skeletal or dental disease and the mutation type and the location of the mutation4,10,14). However, in one study, skeletal disease was more severe in the group with a mutation in the C-terminal half of the protein14). Among patients with a family history of hypophosphatemic rickets, there was a trend toward more severe skeletal disease in patients with truncating mutations4). Hearing defect was correlated with mutations in the beginning fragment of the gene, and dental disease and increased head length was correlated with the mutations in the beginning and the terminal fragment of the gene10).
Without treatment, clinical manifestations begin in the first year of life, and affected children suffer progressive growth failure and bony deformities, such as tibial bowing and frontal bossing14). However, treatment with vitamin D and phosphate resulted in only a partial growth improvement in most cases, and was frequently complicated by hypercalciuria, hypercalcemia, nephrocalcinosis, or hyperparathyroidism10). Despite the fact that the affected individual was treated with vitamin D and phosphate, rickets and growth catch-up were not completely improved in the patient, and finally surgical correction had to be done at 55 months of age. This investigation suggest that early initiated vitamin D and phosphate treatment are not sufficient to correct hypophosphatemic rickets caused by severe loss-of-function mutations, and that an effective therapeutic approach might be required in such circumstances based on new concepts of the molecular mechanism of the disorder. In conclusion, we report a novel de novo splicing mutation in the PHEX gene. The results of this study expand and improve our understanding of the clinical and molecular characteristics and the global pool of patients with sporadic hypophosphatemic rickets.