Introduction
Gonadotropin-releasing hormone (GnRH) secretion is active in infancy and then becomes quiescent in childhood. At the onset of puberty, the reactivation of pulsatile GnRH secretion leads to an increase in the secretion of the gonadotropins and consequent gonadal stimulation1). Central precocious puberty (CPP) is caused by the premature reactivation of the hypothalamic-pituitary-gonadal axis and is usually defined by the development of secondary sexual characteristics before the age of 8 in girls and 9 in boys2,3).
Complex interactions with genetic, nutritional and environmental factors play a crucial role in determining pubertal timing4). Genetic factors are have been considered to have a strong influence on pubertal initiation5). CPP is usually regarded to be idiopathic. de Vries et al.6) mentioned that familial cases accounted for 27.5% (43 out of 156 children) of CPP, and segregation analysis suggested an autosomal dominant transmission with incomplete sex-dependent penetrance.
Numerous studies have been conducted to explore the genetic causes for CPP. The GABRA1, NPY-Y1R, LIN28B, TAC3 and TACR3 genes were considered regarded as potential the cause of CPP, but no mutations associated with CPP were found in these genes7,8,9,10). Recently, kisspeptin (KISS1), kisspeptin receptor (KISS1R), and makorin RING finger protein 3 (MKRN3) genes were found to be responsible for CPP, but a few cases of CPP with these mutations have been reported to date11,12,13).
KISS1 and KISS1R gene
Kisspeptin and its receptor have been considered essential components in pubertal onset. KISS1 encodes kisspeptin, which is a natural ligand that binds to the KiSS-1 receptor (KISS1R), a G-protein coupled receptor14). Several studies have demonstrated that loss of function mutations in KISS1R were detected in idiopathic hypogonadotropic hypogonadism patients and it was suggested that this receptor is a regulator of GnRH secretion15,16). Kisspeptin is shown to activate hypothalamic GnRH secretion, after binding KISS1R in hypothalamic GnRH-neurons14). KISS1 and KISS1R expressions are increase at the onset of puberty14).
In 2008, Teles et al.11) detected a gain-of-function mutation in the KISS1R gene in a girl with CPP. They identified an autosomal dominant p.Arg386Pro mutation. This mutation prolongs the activation of kisspeptin responsive to intracellular signaling and reduces the rate of normal KISS1R desensitization11). In response to kisspeptin stimulation, pulsatile of GnRH secretion increases and induces the initiation of puberty17). Silveira et al.12) reported the p.P74S mutation in KISS1 gene in a boy with very early pubertal development at 12 months of age, and suggested this mutation was associated with higher kisspeptin resistance to degradation, leading to an increased availability of bioactive kisspeptin. However, so far, KISS1 and KISS1R mutation have been very rarely found in patients with CPP18).
MKRN3 gene
In 2013, through whole-exome sequencing analysis, Abreu et al.13) detected MKRN3 gene mutations in 5 of 15 families with CPP. They found that three frameshift mutations (Arg213Glyfs*73, Tyr391*, and Ala162Glyfs*14) were predicted to encode truncated proteins and one missense mutation (Arg365Ser) predicted to disrupt protein function in 5 families originated from North America, Brazil, and Belgium.
The MKRN3 gene is located on chromosome 15q11-q13 in the Prader-Willi syndrome (PWS) critical region19). While studying a PWS critical region in 1999, Jong et al.19) discovered MKRN3 gene. This gene is methylated on the maternal allele, but is unmethylated on the paternal allele. Therefore, the maternal imprinted MKRN3 gene is expressed only from the paternal allele and si silenced from the maternal allele. All affected patients with familial CPP inherited the MKRN3 mutations from their father19). PWS patients usually have delayed or incomplete puberty despite MKRN3 gene deletion. However, rare cases with CPP in PWS have been reported20,21). There is a needs to evaluate genetic factors in 15q11-13 PWS critical region as related to pubertal timing.
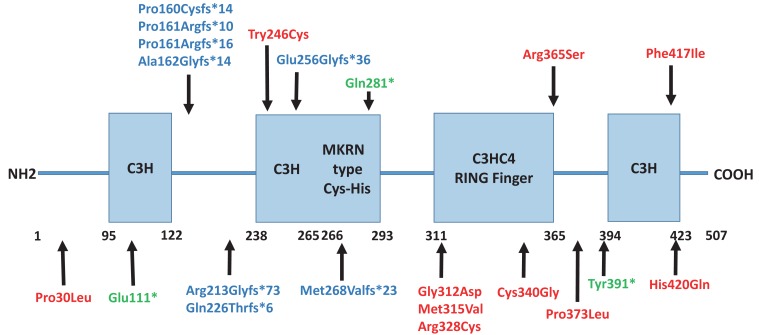
The MKRN3 protein has 2 N-terminal C3H zinc finger motifs, 1 MKRN specific Cys-His domain, 1 C3HC4 RING zinc finger motif, and 1 C-terminal C3H zinc finger motif19). Until now, 21 MKRN3 mutations have been described including 8 frameshift mutations, 10 missense mutations and 3 nonsense mutations13,22,23,24,25,26,27,28,29) (Fig. 1). Abreu et al.13) suggested that the function of MKRN3 gene relevant to pubertal initiation include an inhibiting effect on the pubertal pulsatile GnRH secretion. Their study showed that hypothalamic Mkrn3 mRNA levels were increased in the arcuate nucleus of male and female mice at a young age and decreased immediately before puberty. They suggested they mentioned the Mkrn3 mRNA expression pattern is associated with an inhibitory effect on onset of puberty. Ojeda and Lomniczi30) also suggested that MKRN3 inhibits the downstream activators of hypothalamic GnRH secretion, such as KISS1. MKRN3 deficiency due to a loss of function mutation leads to the withdrawal of hypothalamic inhibition, which releases and pulsatile GnRH secretion, and therefore precipitates the early onset of puberty22). In a study by Hagen et al.31), the authors ascertained undetectable or low MKRN3 levels in patients with early onset of puberty, and MKRN3 levels were negatively correlated with gonadotropins in serum from prepubertal girls.
Settas et al.22) demonstrated that a novel heterozygous missense MKRN3 mutation (p.Cys340Gly) detected in 2 affected Greek sibling. The p.Cys340Gly mutation was predicted to disrupt the protein function in silico analysis. These 2 patients, a girl with CPP and a boy with early puberty, had inherited the mutated MKRN3 gene from their asymptomatic carrier father and paternal grandmother. Macedo et al.23) reported five novel heterozygous mutations in eight unrelated Brazilian patients with sporadic CPP. These mutations were composed of 4 frameshift mutations, predicted to encode truncated proteins, and 1 missense mutation (p.Phe417Ile), which were all informed of paternally inheritance in segregation analysis. de Vries et al.24) identified a novel missense mutation (p.His420Gln) predicted to disturb RNA binding in the MKRN3 gene. Schreiner et al.25) identified 2 heterozygous mutations (a previous reported variant, p.Ala162Glyfs*14 and a novel mutation, p.Glu111*) in the MKRN3 gene in 2 German families with CPP.
All patients with loss of function mutations in MKRN3 gene show have typical clinical and hormonal finding of CPP, including breast or testicular enlargement, advanced bone age, accelerated growth velocity, and stimulated luteinizing hormone levels elevation13,22,23). Very few patients with MKRN3 mutations had mild syndromic features. Two related patients had esotropia13) and a girl had mild nonspecific features, including a high arched palate, clinodactyly, dental abnormalities and hyperlordosis23). Apart from these, no other signs were evident in CPP patients with MKRN3 mutations including features of PWS13,23). MKRN3 mutations affect both genders equally but seem to affect girls more severely in the onset age for puberty13,22). The reason for this gender difference is not yet clear. Between CPP patients with MKRN3 mutation and those without MKRN3 mutation, the age of pubertal initiation does not differ significantly23). However, another study has reported the girls with MKRN3 mutations are significantly younger at puberty onset than those without mutations27). Response to treatment with depot GnRH agonists are good in CPP patients with MKRN3 defects13,23,27).
Conclusions
Pubertal timing is determined by interaction between inhibitory effects and stimulatory effects of hypothalamic-pituitary-gonadal axis. It is generally accepted that the MKRN3 gene is the most frequently known genetic cause of pubertal initiation. However, the exact mechanism involved in pubertal development are still not well understood. Larger studies on the clinical features of CPP patients with MKRN3 mutations are required and an ongoing effort to find other factors associated with the puberty should be maintained.