Highlights
Ā· Thyroid hormone resistance (RTH) is a syndrome characterized by reduced target organ sensitivity to thyroid hormone,which present a various clinical phenotypes, often leading to misdiagnosis. We report the 4-year clinical course of a boy who diagnosed with RTH due to novel variant mutation in THRĪ².
Introduction
Thyroid hormone (TH) resistance (RTH) is characterized by reduced target tissue sensitivity to TH, elevated serum TH level, and failure to suppress pituitary thyroid-stimulating hormone (TSH) secretion. RTH was first described in 1967 [1], and its prevalence is 1:40 ,000 [2]. RTH is caused by a defect in THRĪ±- and THRĪ²-encoded thyroid hormone receptors (THRs). Approximately 90% of RTH patients carry a typically autosomal-dominant THRĪ² mutation; at least 236 mutations have been identified in 805 families [3]. THR isoforms are distributed in various tissues and present varying degrees of hormonal resistance depending on the relative levels of gene expression, resulting in varying symptoms. A patient may show signs and symptoms of hypothyroidism in one tissue and suggestive thyrotoxicosis in others. Some patients might be asymptomatic and in a euthyroid state [3,4], suggesting that RTH can be easily misdiagnosed based on symptoms or thyroid function tests. Genetic tests are diagnostic tools that can reduce unnecessary drug administration and promote individualized treatment. Here, we report the 4-year clinical course in a Korean boy with RTH with a novel THRĪ² variant, c.993T>G (p.Asn331Lys).
Case report
A 12-month-old boy, referred to the Department of Pediatric Endocrinology for abnormal thyroid function test results, presented with hyperthyroxinemia and an inappropriately increased TSH level. He was a first-born, delivered by cesarean section at 28 weeks 2 days of gestation because of premature membrane rupture, and had no family history of goiter or autoimmune thyroid disease (AITD). His birth weight, length, and head circumference were 1,100 g (-0.08 standard deviation score [SDS]), 35 cm (-0.76 SDS), and 27 cm (0.78 SDS), respectively, and his 1- and 5-min Apgar scores were 7 and 8, respectively.
After birth, prophylactic surfactant replacement therapy and an invasive ventilator were initiated. A physical examination at 6 days after birth did not indicate goiter; however, a routine venipuncture thyroid function test revealed a TSH level of 16.67 Ī¼IU/mL (reference range, 0.35ā4.94 Ī¼IU/mL) and a free T4 (fT4) level of 2.47 ng/dL (reference range, 0.7ā1.48 ng/dL). The pulse rate was 154ā166 per minute (reference rate, 140ā184 per minute). Total bilirubin and direct bilirubin were 7.4 mg/dL and 0.78 mg/dL, respectively. Although venous TSH concentration was <20 Ī¼IU/mL, levothyroxine (10 Ī¼g/kg/day) was initiated. At 49 days after birth, TSH and fT4 levels were 75.9 Ī¼IU/mL and 1.2 ng/dL, respectively, and levothyroxine dose was increased to 30 Ī¼g/kg/day. At 12 months, an elevated fT4 (3.22 ng/dL) and an inappropriately high TSH level (2.47 Ī¼IU/mL) continued, and the patient was referred to the Department of Pediatric Endocrinology. He had no history of heat intolerance, excessive sweating, weight loss, constipation, or developmental delay on admission. His age-corrected height was 73 cm (-0.12 SDS), weight 7.42 kg (-1.92 SDS), blood pressure 90/60 mmHg (reference range, 80ā100/55ā65 mmHg), pulse rate 84 beats/min (reference range, 80ā120 beats/min), and body temperature was 36.7ā, which were all within the normal ranges. On physical examination, goiter, thrill over the thyroid gland, and exophthalmos were not observed. His developmental quotient by Korean infant and toddler development-screening tests was within the normal range for his corrected age. His thyroid function test revealed serum fT4, T3, and TSH levels of 2.89 ng/dL, 1.9 ng/mL, and 5.33 Ī¼IU/mL, respectively. Normal levels of total cholesterol (137 mg/dL; reference range, <200 mg/dL), triglycerides (63.8 mg/dL; reference range, <130 mg/dL), highdensity lipoprotein cholesterol (46.9 mg/dL; reference range, >40 mg/dL), and low-density lipoprotein cholesterol (77.3 mg/dL; reference range, <100 mg/dL) were noted. Hemoglobin concentration was 13.9 g/dL (reference range, 11.3ā14.1 g/dL). The results of antithyroglobulin antibody and TSH receptor antibody tests were negative, whereas that of antimicrosomal antibody was positive (12.83 IU/mL; reference range, <12 IU/mL). Free alpha subunit (0.3; reference range, <0.7 ng/mL) and sex hormone-binding globulin (SHBG; 123.8 nmol/L; reference range, 41.5ā150 nmol/L), which are associated with TSHsecreting tumor, were not elevated.
The thyrotropin-releasing hormone stimulation test showed an exaggerated TSH response (TSH at baseline, 7.02 Ī¼IU/mL; at 30 minutes, 88.55 Ī¼IU/mL; at 60 minutes, 73.4 Ī¼IU/mL; at 90 minutes, 79.27 Ī¼IU/mL; at 120 minutes, 64.41 Ī¼IU/mL). Magnetic resonance brain imaging revealed no evidence of pituitary adenoma; however, Rathke cleft cyst was found (Fig. 1). Thyroid ultrasonography showed normal size, contour, parenchymal echogenicity, and vascularity without nodules. The 99m Technetium thyroid scan revealed increased uptake in both normal-sized thyroid lobes.
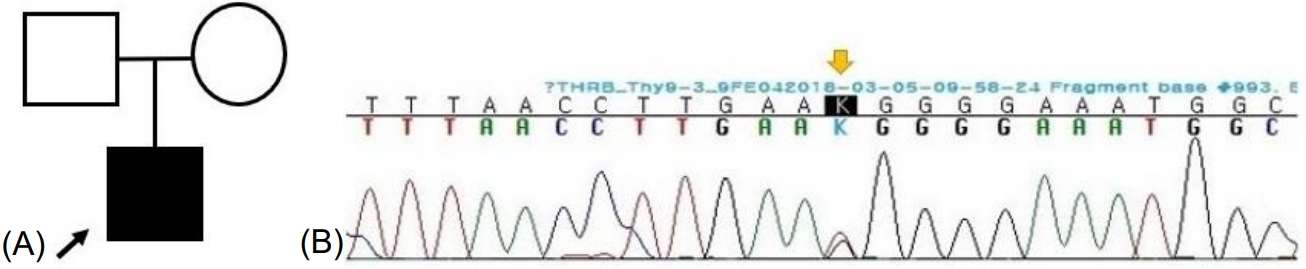
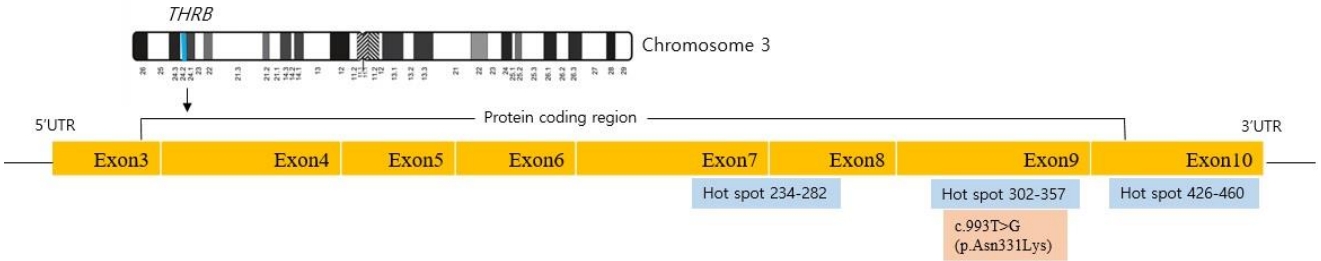
Owing to the possibility of RTH, a targeted next-generation sequencing panel test associated with hypothyroidism that included THRĪ±, THRĪ², SLC16A2, DUOX2, DUOXA2, FOXE1, GNAS, HESX1, IYD, LHX3, NKX2-1, NKX2-5, PAX8, POU1F1, PROP1, SLC26A4, SLC5A5, TG, TPO, TRH, TRHR, TSHB, and TSHR was conducted after obtaining informed consent from his parents. Genomic DNA was extracted from the peripheral blood of the patient. The test revealed a preheterozygote c.993T>G (p.Asn331Lys) mutation in THRĪ². (Figs. 2, 3) An in silico analysis (MutationTaster, PolyPhen-2, and SIFT) predicted this mutation to be deleterious (PP3). The variant was novel (http://www.hgmd.cf.ac.uk, access date: November 15, 2021.) and located at a mutational "hot spot" (PM1) that codes for the T3-binding domain [5]. According to the data from the 1000 Genomes Browser (2,504 individuals, http://phase3browser.1000genomes.org) and ExAC (60,706 individuals, http://exac.broadinstitute.org/) databases and gnomAD v2.1.1 (https://gnomad.broadinstitute.org), this variant was absent in control individuals (PM2). Moreover, a missense pathogenic mutation including c.993T>G (p.Asn331Lys) has been reported at the same site (PM5) [6]. Therefore, the variant was classified as likely pathogenic according to the ACMG/AMP guidelines (ā„3 moderate and 1 supporting). The patient had no siblings, and the thyroid function of his parents was normal; for economic reasons, the parents did not agree to undergo genetic tests on themselves.
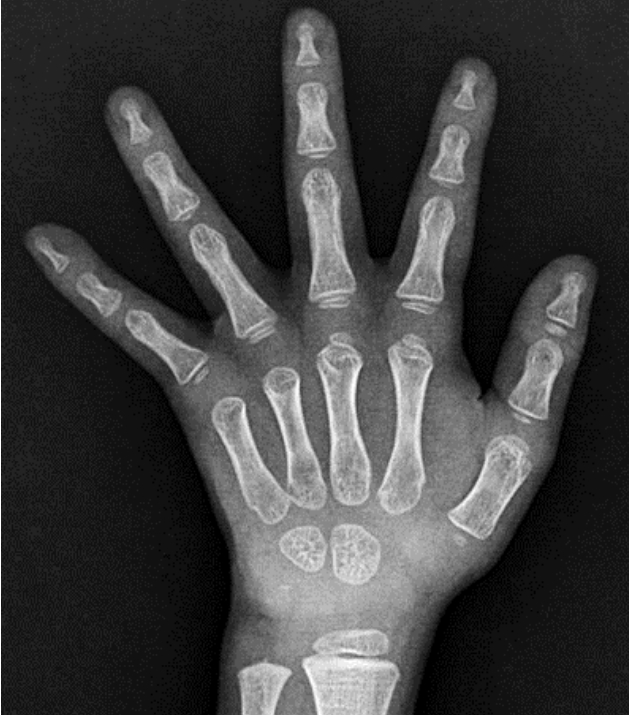
During the administration of levothyroxine, sinus tachycardia (pulse rate 88ā131 beats/min) was observed in the outpatient clinic; however, heat intolerance or hyperactivity was not. Finally, the boy was diagnosed with RTH and transferred to another hospital, closer to his hometown, where the physician restarted levothyroxine to maintain a TSH level < 5 Ī¼IU/mL. The dose was gradually reduced, and the drug was discontinued at 3 years. However, after 3 months of levothyroxine discontinuation, TSH level had increased to 10.2 Ī¼IU/mL without goiter, and 2ā3 ug/kg/day levothyroxine was readministered. At 4 years 8 months, his TH level was elevated with unsuppressed TSH level (TSH, 5.61 Ī¼IU/mL; fT4, 3.03 ng/dL; T3, 1.9 ng/mL) (Table 1). At that time, the patient showed insufficient growth and weight gain. His height was 100.2 cm (-1.73 SDS), weight was 12.9 kg (-3.38 SDS), and body mass index (BMI) was 12.8 kg/m2 (-2.73 SDS); however, his development was normal. His bone development was delayed with respect to his chronological age (Fig. 4).
Discussion
RTH exhibits various clinical manifestations and is often misdiagnosed as hypo- or hyperthyroidism, resulting in ineffective drug or surgical treatment. Ablative treatment or surgical resection under the diagnosis of hyperthyroidism or goiter requires TH administration, often in supraphysiological doses [7]. Differential diagnoses include TSH-secreting pituitary adenoma, familial dysalbuminemic hyperthyroxinemia, and assay interference. RTH should be suspected if the following conditions are present: (1) absence of elevated Ī± pituitary glycoprotein-subunit serum concentration, (2) normal or increased TSH response after TRH administration, (3) family history of RTH, (4) absence of elevated serum SHBG concentration, reflecting a euthyroid state, (5) suppression of serum TSH level with supraphysiological doses of L-T3, and (6) identification of THRĪ± and THRĪ² mutations by genetic testing [8].
The molecular structures and sequences of THRĪ± and THRĪ² are similar; 4 isomers are denoted as THR-Ī±1, -Ī±2, -Ī²1, and -Ī²2 [9]. THRĪ±1 is expressed primarily in the heart, bone, and brain, whereas THRĪ±2 is widely distributed but is unable to bind to hormones. THRĪ²1 is predominantly expressed in the liver and kidney, and THRĪ²2 is highly expressed in the pituitary gland and hypothalamus [10,11]. Therefore, symptoms of TH deficiency and excess could coexist in different tissues of the same patient. For example, growth retardation and learning difficulty (suggestive of hypothyroidism) may coexist with weight loss, osteoporosis, and tachycardia (typical of thyrotoxicosis) [4]. Tachycardia may be explained by high THRĪ±1 concentration in heart cells [7].
Most THRĪ² mutations are concentrated in 3 "hot spots" between exons 7 and 10 (234ā282, 310ā353, 429ā461) [12]; in the present case, c.993T>G (p.Asn331Lys) was identified. To our knowledge, only 3 patients (patients 1, 2, and 3) with RTH who had mutation at codon 331 in exon 9 have been previously reported (Table 2) [5,6,13]. Patient 4 was the patient of the present report. Patients 1 and 2 inherited THRĪ² mutations from their mothers, and the other exhibited de novo mutation. On physical examination, goiter was found in 2 of the 3 patients. Tachycardia was investigated in patients 1 and 4, and heart rate was within the normal range in both. Anthropometric parameters were measured for patients 1, 3, and 4. Height and BMI standard deviations varied from 0.9 to -1.75 and 0 to -2.73, respectively. Measurements in patients 1 and 4 indicated delayed bone age compared with chronologic age. TSH, fT4, and T3 levels were 2.2ā15.2 Ī¼IU/mL, 2.2ā2.89 ng/dL, and 1.9ā8 ng/mL, respectively. Antimicrosomal antibodies were found in the 3 tested patients. Antithyroglobulin antibodies were found in 1 of 2 patients who underwent the examination. Long-term outcome was reported in patient 1, who experienced late puberty, no learning difficulty, and a normal adult life with 0.02 mg/kg/day triiodothyroacetic acid treatment.
In most patients with RTH, high level of circulating TH might compensate for partial tissue resistance. Therefore, RTH treatment should be tailored to correct the coexistence of hypo- and hyperthyroidism in various tissues. Selective beta-blockers can be considered for patients with tachycardia or tremor. Triiodothyronine (L-T3) treatment may benefit goiter, hyperactivity, and mental clouding [3]. Administering supraphysiologic L-T3 (250 Ī¼g) every other day effectively reduced goiter size in large symptomatic goiter cases [14]. Levothyroxine treatment is indicated when RTH coexists with thyroid dysgenesis, such as in ectopic thyroid glands or hypothyroidism due to autoimmune thyroiditis [15]. Supraphysiologic levothyroxine is often needed to maintain serum TSH at the lowest tolerable level [3]. Additionally, these symptoms and basal metabolic rate, nitrogen balance, and serum SHBG should be monitored at each dose escalation [16].
Indeed, RTH patients had a higher frequency of AITD, in which elevated TSH stimulates intrathyroidal lymphocytes, which results in the production of proinflammatory cytokines and the destruction of thyrocytes [17]. Meanwhile, patients with RTH due to THRĪ² mutation have an approximately 2-fold higher risk of AITD compared to unaffected relatives, but the prevalence of thyroid autoantibodies with aging is not affected by genotype [18]. In the present case, antimicrosomal antibody was positive at 12 months of age, but it was just above the normal range. Normal radiographic findings were observed on thyroid scan and ultrasound. Serological evidence of thyroid autoimmunity including antithyroglobulin antibody, TSH receptor antibody, and antimicrosomal antibody were not observed around age 3 or 4. For proper management of this patient, further regular evaluation for thyroid autoimmunity is needed.
Treatment guidelines for neonatal patients with RTH and congenital hypothyroidism are lacking. As previously mentioned, L-T3 treatment could be an option for patients with RTH; however, L-T3 has not been reported administered for congenital hypothyroidism [19]. Therefore, a clinician can consider levothyroxine treatment. The goal of treatment with supraphysiologic levothyroxine doses is to return serum TSH level to near normal while monitoring cognitive development, bone maturation, and growth [3]. To maintain TSH below 5 Ī¼IU/mL, levothyroxine (up to 350 Ī¼g/day) was administered to a young female patient presenting RTH combined with ectopic thyroid glands [20]; however, prolonged treatment with levothyroxine after age 3 resulted in poor weight gain.
In conclusion, RTH does not exhibit specific clinical features but may be associated with tachycardia, goiter, growth retardation, or other symptoms. Due to its heterogeneous nature, each patient should be individually evaluated and treated. If RTH is suspected, clinicians should consider either a thyroid scan or ultrasound to check for thyroid dysgenesis or laboratory examinations to determine hypothyroidism associated with autoimmune thyroiditis. Long-term follow-up data and a detailed diagnosis and treatment strategy for RTH are necessary.