Highlights
┬Ę The CYP21A2 gene is located in the major histocompatibility complex class III region on chromosome 6p21.3, approximately 30 kb apart from its pseudogene, CYP21A1P.
┬Ę Molecular diagnosis of 21-hydroxylase deficiency (21-OHD) is challenging because of high sequence homology between CYP21A2 and CYP21A1P, with approximately 98% homology at the exon level and 96% homology at the intron level.
┬Ę The phenotype of 21-OHD varies according to the degree of residual enzyme activity.
┬Ę Genetic diagnosis is helpful in guiding appropriate management and predicting the phenotype and prognosis of patients with 21-OHD.
Introduction
Congenital adrenal hyperplasia (CAH) is a heterogeneous group of enzyme deficiencies in the steroidogenic pathway of the adrenal cortex that result in impaired synthesis of cortisol and aldosterone by the adrenal glands [1]. Impaired cortisol synthesis causes prolonged elevation of adrenocorticotropic hormone, which leads to overstimulation of the adrenal cortex and subsequent adrenal gland hyperplasia. More than 95% of patients with CAH have steroid 21-hydroxylase deficiency (21-OHD), which is the lack of an enzyme encoded by the CYP21A2 gene [2]. This condition is classified into classic and nonclassic forms based on the residual enzyme activity. The classic form is defined by severely reduced or absent enzyme activity, and it is subdivided into the salt-wasting (SW) form with little or no enzyme activity and the simple virilizing (SV) form with 1% to 5% of residual enzyme activity. Nonclassical CAH (NCCAH) is the mildest form of CAH; it does not cause virilization or a life-threatening adrenal crisis.
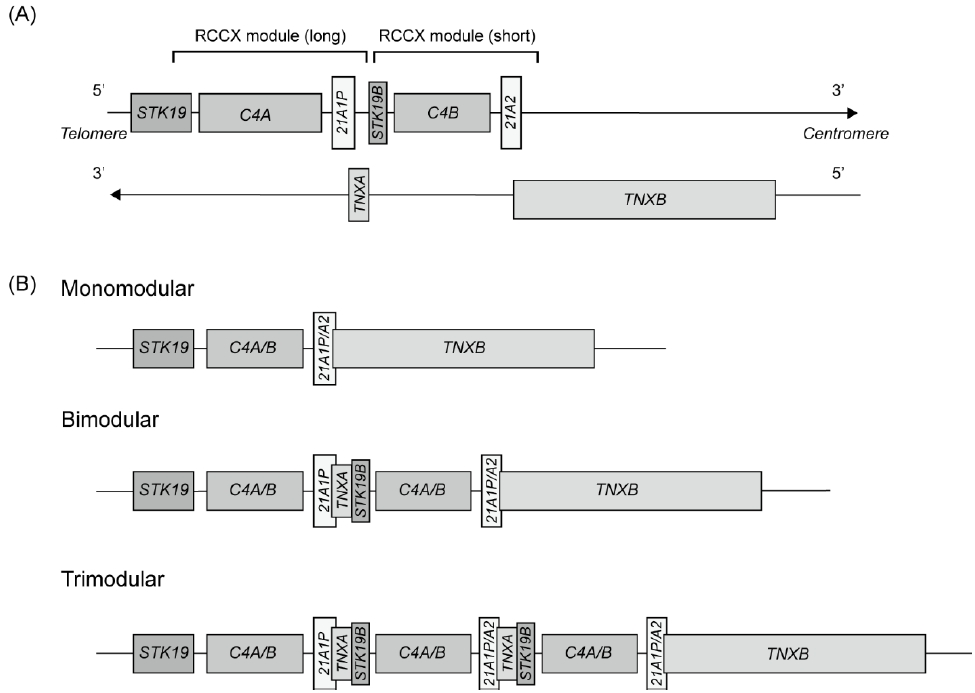
The CYP21A2 gene spans 3.35 kb and consists of 10 exons [3,4]. It is located in the major histocompatibility complex class III region of chromosome 6p21.3, and tandem repeats of the active genes and its pseudogenes are aligned within the RCCX module. The tandem repeats often cause complex genomic recombination events between the CYP21A2 and CYP21A1P genes, which are approximately 30 kb apart.
This review describes the molecular mechanism of 21-OHD, its genotype-phenotype correlations, the extended phenotypes of CAH-X syndrome, and genetic testing strategies. Many studies have reported inconsistent nomenclature of mutations in CYP21A2 that result from numbering the initiation codon as 0. In this review, mutations are described according to nomenclature guidelines from the Human Genome Variation Society (https://varnomen.hgvs.org/), in which nucleotide numbering begins with the translation initiation codon (ATG) as the first codon number, and subsequent nucleotides are numbered from the 5' to 3' direction [5].
Genetics of the CYP21A2 gene and molecular mechanisms of 21-OHD
1. Genetics of CYP21A2 and organization of the RCCX module
The CYP21A2 gene is located in a complex organization of genes known as the RCCX module, which is a tandem repeat structure containing a series of functional and nonfunctional genes: serine/threonine kinase 19 (STK19, formerly RP), complement 4 (C4), steroid 21-hydroxylase (CYP21), and tenascin-X (TNX) (Fig. 1A) [3,6]. The RCCX module consists of 2 segments, the short and long modules [7]. The short module contains STK19B, C4B, CYP21A2, and part of TNXB. The long module consists of part of the STK19 gene, the full length of the C4A gene, the CYP21A1P gene, and the TNXA gene. Because of the high sequence homology between the active gene and the pseudogene, microconversions and large rearrangements occur through intergenic recombination [1].
The STK19 gene, which encodes a nuclear serine/threonine kinase protein, was recently proposed to be a regulator of NRAS activity [8]. STK19 phosphorylates NRAS, which facilitates interactions between NRAS and its downstream effectors, increasing the activation of the mitogen-activated protein kinase cascade. C4A (active gene) and C4B (pseudogene), each contain 41 exons and are highly homologous and essential elements of the humoral immune response. The size of the human C4 gene varies from 16 kb to 22 kb due to the integration of the endogenous retroviral sequence (6.7 kb), human endogenous retrovirus-K (HERV-K), in intron 9 of the long C4 gene [9]. Approximately 65% of human C4 gene contains HERV-K viral integration. Although no evidence links HERV-K to human disease, aberrant expression of these retroviral elements is associated with the development of autoimmune diseases [10,11]. Thus, C4 deficiency correlates strongly with autoimmune diseases such as systemic lupus erythematosus [12]. The transcription start site (5'-end) of the CYP21A1P and CYP21A2 genes is located 2,466 bp downstream of both C4A and C4B [13,14].
The CYP21A2 gene encodes 21-hydroxylase, which plays a critical role in the synthesis of the 2 principal steroid hormones, aldosterone and cortisol. CYP21A2 and its pseudogene CYP21A1P share a high degree of sequence homology, with 98% sequence identity in the coding region and 96% in the intronic region [4,15]. The pseudogene is inactivated by several pathogenic variants that prevent the synthesis of a functional protein.
The TNXA and TNXB genes have an opposite transcriptional direction compared with other cluster genes [6,16]. The 3'-ends of the CYP21 genes overlap with the last exons of TNXA and TNXB (Fig. 1A) [6]. The TNXB gene encodes an extracellular matrix glycoprotein, TNX, that consists of 44 exons spanning 68.2 kb. Defects in TNXB cause Ehlers-Danlos syndrome (EDS) [16]. In contrast, TNXA is a truncated pseudogene spanning 4.5 kb and is homologous to TNXB from exons 32 to 44.
A germline copy number variation (CNV) is a fragment of DNA with variable copies that result from duplications or deletions during evolution. Multi-allelic CNVs are genomic segments that exhibit a variable number of copies among individuals. They can affect gene expression, protein function, and phenotypic traits, thereby contributing to genetic and phenotypic heterogeneity. The RCCX module is one of the most complex CNV loci in humans [3]. In Caucasians, RCCX CNV alleles typically have monomodular, bimodular, or trimodular segments, with a prevalence of approximately 17%, 69%, and 14%, respectively (Fig. 1B) [10]. The genetic variations in RCCX CNVs make it difficult to accurately characterize molecular defects in CYP21A2.
2. Molecular mechanisms of genetic defects in CYP21A2
1) Point mutations by microconversion
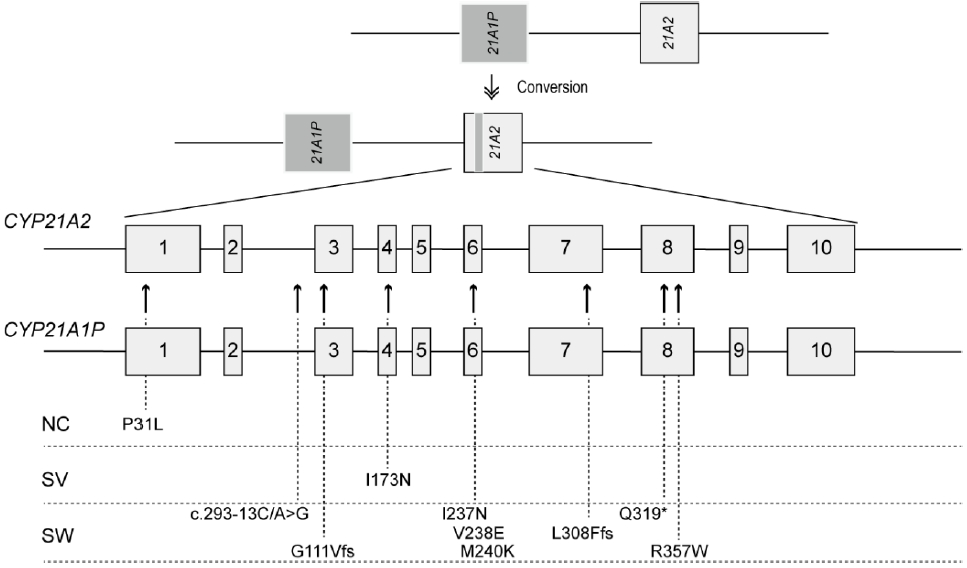
As mentioned above, the molecular diagnosis of 21-OHD is challenging because of high sequence homology between CYP21A2 and CYP21A1P. They differ by only 65 nucleotides in their coding and intronic regions [4]. Microconversion of sequence variants from CYP21A1P to CYP21A2 is responsible for nonfunctional protein synthesis in 70%ŌĆō75% of cases (Fig. 2) [2]. Deleterious variants are transferred by small conversions from the pseudogene during meiosis. Microconversion events are caused by major recurrent variants: a splicing mutation (c.293-13A/C>G, also known as I2G), an 8-bp deletion in exon 3 (p.G111Vfs), one nucleotide insertion in exon 7 (p.L308Ffs), 1 nonsense mutation (p.Q319*), 3 missense mutations (p.P31L, p.I173N, and p.R357W), and 1 cluster conversion (p.I237N, p.V238E, and p.M240K) (Fig. 2) [17,18].
2) Large gene rearrangement (chimeric CYP21A1P/CYP21A2 and TNXA/TNXB) by unequal meiotic crossover
In the remaining 25%ŌĆō30% of cases, large gene rearrangements due to unequal crossover during meiosis result in large deletion, duplications, or other contiguous gene deletions [2]. Chimeric CYP21A1P/CYP21A2 genes occur by homologous recombination between these 2 genes. A 26- or 32-kb deletion (depending on the size of C4B) in the region involved in the 3'-end of CYP21A1P, TNXA, and C4B and the 5'-end of CYP21A2 creates a single, nonfunctional chimeric gene (Fig 3A). To date, 9 different chimeric CYP21A1P/CYP21A2 genes have been characterized based on the chimeric junction site (Fig. 3A) [19]. These chimeric genes have been classified into classic and attenuated forms, depending on whether the junction site is upstream or downstream of the I2G mutation [19]. Seven chimeras (CH1, CH2, CH3, CH5, CH6, CH7, and CH8) carrying this splicing mutation (I2G) are associated with a severe SW phenotype [18,19]. In contrast, 2 chimeras (CH4 and CH9) harboring the promoter of CYP21A1P and the p.P31L mutation are associated with an attenuated phenotype [19].
3. Genotype-phenotype correlations for mutations in CYP21A2
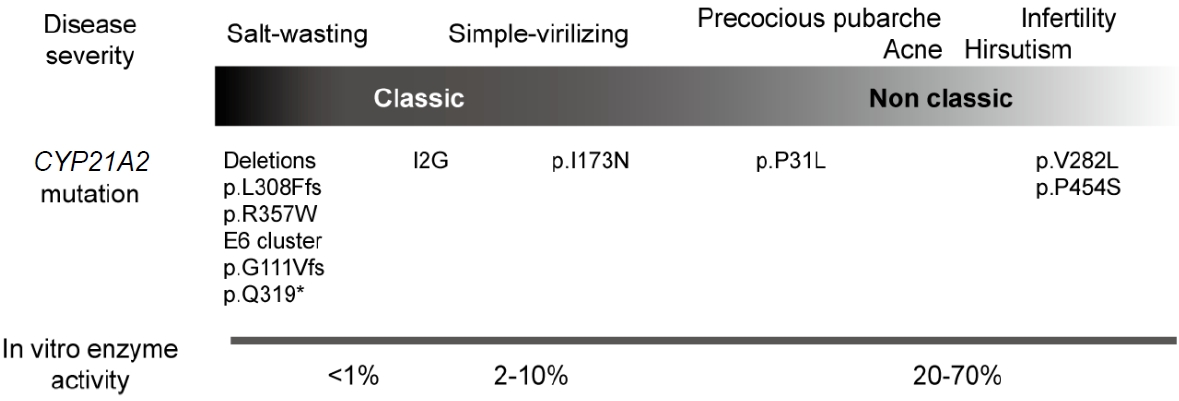
Common major mutations correlate with phenotypes that reflect varying degrees of residual enzyme activity (Fig. 4). Mutations in CYP21A2 are categorized into 4 groups (0, A, B, and C) based on in vitro data according to the level of enzyme activity [23]. Group 0 is caused by null mutations, such as a large deletion, p.L308Ffs, p.Q319*, and p.R357W, that leave no enzyme activity. Group A consists of mutations that cause severely impaired enzyme activity, such as a homozygous or compound heterozygous I2G mutation. Group B reflects either homozygous or compound heterozygous for the p.I173N mutation with group 0 or A variants. Group C consists of milder homozygous or compound heterozygous mutations, such as p.V282L, p.P454S, and p.P31L, which retain 20%ŌĆō60% of residual enzyme activity [1].
Because most patients with 21-OHD harbor compound heterozygous mutations, the allele with the less severe mutation tends to have a greater influence on the clinical phenotype. Severe genotypes (group 0 and A) correlate well with the SW form, ranging from 91%ŌĆō97%; however, this relationship is less strong for the intermediate severity groups (group B and C), which range from 45%ŌĆō57% [24]. For example, patients carrying the p.P31L mutation predominantly have NCCAH, but that mutation has also been reported in the SV type [1,24,25]. This genotype-phenotype discordance could be explained by a genetic modifier. Combined variants of p.P31L and 4 single nucleotide polymorphisms in the promoter region (c.-126C>T, c.-113G>A, c.-110T>C, and c.-103A>G) have been shown to reduce transcriptional activity by 5-fold and enzyme activity by less than 6% [26]. This reduction leads to a more severe phenotype than would be expected on the basis of the genotype alone. Because little research on genetic modifiers in CAH has been done, further studies are needed to fully understand their role. Therefore, genotype-phenotype correlations are not always absolute, and clinical management should be based on clinical features and hormonal data rather than the genotype [1,25,27].
4. Mutation spectrum of CYP21A2 according to ethnicity
The worldwide incidence of classic CAH, based on neonatal screening, ranges from 1:14,000 to 1:18,000 births [28]. The prevalence of the major mutations varies by ethnicity and geographic region (Table 1). Deletion/conversion, I2G, and p.I173N mutations are the most common in most populations [1]. However, the p.V282L mutation is quite common in Ashkenazi Jews with 21-OHD, and it has rarely been observed in Asians [27]. The I2G and p.V282L mutations are more prevalent in the Middle Eastern population than elsewhere, and the p.Q319* mutation is the most common in the Tunisian population [29,30]. In Korean patients, large deletions are the most common mutation, followed by the I2G and p.I173N [31]. This ethnic diversity and specificity of CYP21A2 mutations could facilitate targeted genetic screening and improve the efficiency of 21-OHD diagnosis.
CAH-X syndrome
Deletions affecting both the CYP21A2 and TNXB genes result in a contiguous gene deletion syndrome known as CAH-X syndrome, which is characterized by a hypermobile form of EDS [14,32]. The tenascins are widely-expressed extracellular matrix proteins, and TNX is expressed in the connective tissues, such as skeletal muscle, heart, and blood vessels [16,32]. The role of TNX has been confirmed by Tnx-knockout mice, which have reduced collagen content and connective tissue architecture, suggesting that TNX plays an essential role in regulating collagen deposition in connective tissues [33]. The incidence of CAH-X syndrome is estimated to be 7%ŌĆō15% of patients with 21-OHD [34-36]. However, the exact prevalence of the condition remains unknown because it is often underdiagnosed due to the variability of the phenotype and lack of awareness among physicians.
The TNXA gene is a partially duplicated gene segment that shares a high degree of sequence homology with TNXB from intron 32 to exon 45. TNXA is truncated at the 5' end and harbors a 120-bp deletion causing a frameshift and premature termination of translation [37]. A chimeric TNXA/TNXB gene is caused by unequal gene crossover in the RCCX module, resulting in the complete deletion of the STK19B-C4B-CYP21A2 genes with a fraction of the TNXA and TNXB genes (Fig. 3B) [38]. Three different TNXA/TNXB chimeras have been reported based on the location of the junction site [39]. CAH-X CH1 is caused by a 120 bp deletion in exon 35, which can be detected by a multiplex-ligation dependent probe amplification (MLPA) analysis. Because exons 32ŌĆō34 and exons 36ŌĆō44 are highly homologous, CNV detection in an MLPA analysis is difficult. CAH-X CH2 is characterized by an intact exon 35 and the p.C4058W mutation in exon 40. CAH-X CH3 has a cluster of 3 pseudogene-derived variants: p.R4073H in exon 41, p.D4172N in exon 43, and p.S4175N in exon 43 [38].
To date, the clinical features of more than 50 patients with CAH-X syndrome have been described [34-36,38,40]. Overall, skin laxity and musculoskeletal features, such as joint hypermobility and subluxation, are common. Chronic arthralgia is more prominent in adult patients with CAH-X syndrome than in children. Cardiac abnormalities such as septal defects, cardiac chamber enlargement, or great vessel enlargement occur in about 25% of patients with this condition. These patients also have gastrointestinal abnormalities, including gastroesophageal reflux, hernia, and rectal prolapse. Other findings include early-onset osteoarthritis, scoliosis, pectus excavatum, osteoporosis, bifid or elongated uvula, easy bruising, and delayed wound healing. Patients with biallelic deletion of TNXB have more severe clinical manifestations than those with monoallelic deletion [35].
Molecular genetic testing in patients with 21-OHD
Molecular genetic testing for 21-OHD is a complex process due to the genomic structure of CYP21A2 and the surrounding region. Various disease-causing mutations include point mutations, large gene rearrangements, multiple mutations in cis, and CNVs of variable sizes. Large gene rearrangements can be detected by Southern blot analyses, real-time quantitative polymerase chain reaction (PCR), and MLPA analyses. Currently, Sanger sequencing combined with an MLPA analysis is commonly used and can detect most mutations.
1. Southern blot analysis and quantitative PCR
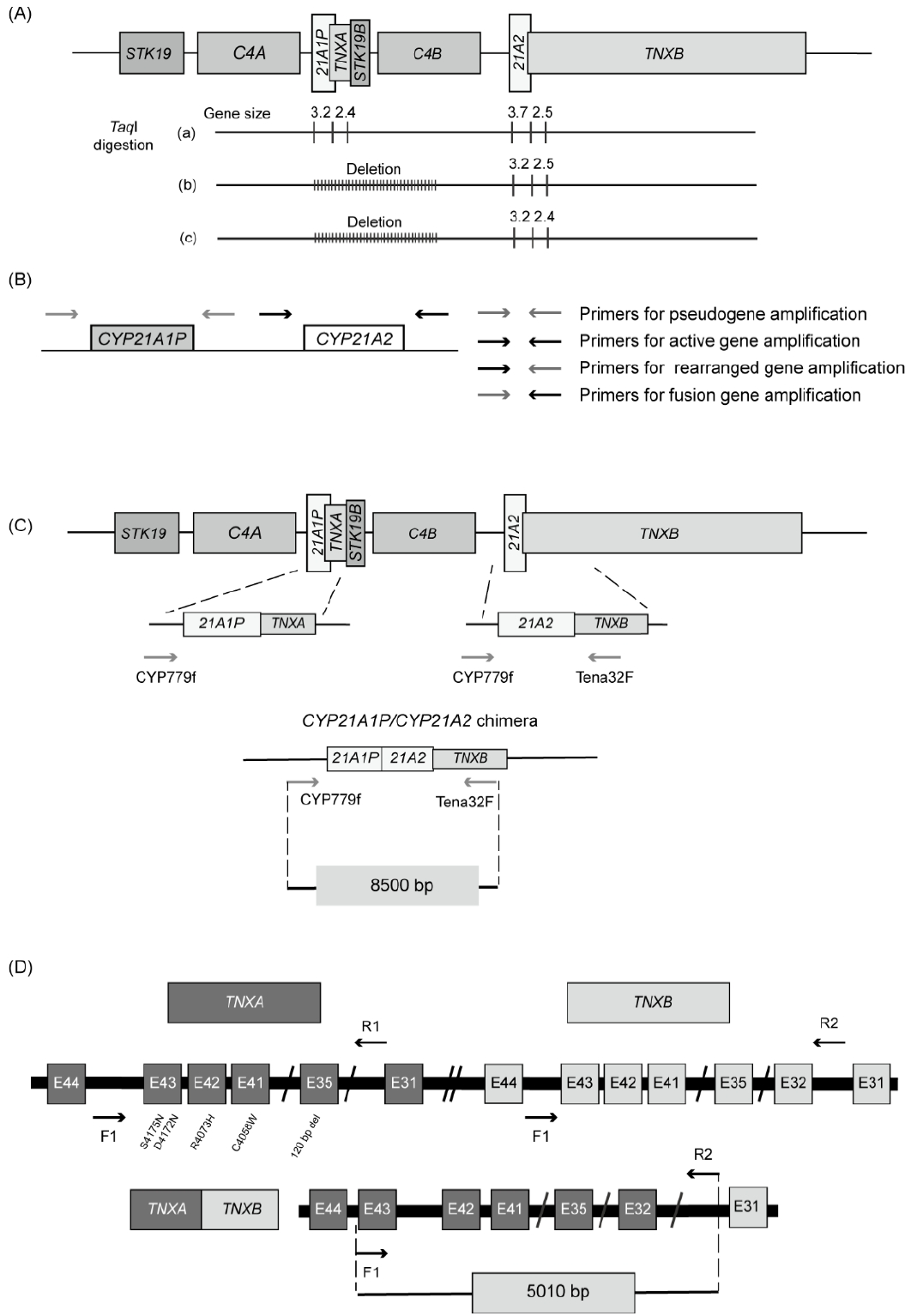
Southern blot analyses were originally used to detect large gene deletions or duplications. This method uses restriction endonucleases that recognize short DNA sequences and cleave double-stranded DNA. The most commonly used restriction endonuclease is TaqI, which recognizes the palindromic DNA sequence of 5'-T^CGA-3', where the caret symbol (^) indicates the cleavage site. This enzyme produces 3.7- and 2.5-kb fragments of intact CYP21A2 and the partial TNXB gene, respectively, and 3.2- and 2.4-kb fragments of CYP21A1P and the partial TNXA gene, respectively (Fig. 5A) [41]. The 3.2- and 3.7-kb fragments are indicative of the CYP21A1P and CYP21A2 genes by their size. Southern blot analyses show that the 3.2-kb fragment without the 3.7-kb fragment represents deletion of the CYP21A2 gene (Fig. 5A). However, this method is time-consuming and requires large amounts of DNA.
Real-time, quantitative PCR is a rapid and sensitive method for detecting gene deletions and duplications. This method uses CYP21A2-specific and internal control gene primer sets and measures the threshold cycle (Ct) values of both the target and control genes. To assess the CYP21A2 copy number, the difference in Ct for CYP21A2 and the internal control gene is calculated and plotted against the logarithm of the CYP21A2/internal control ratio (%). However, some discrepancies with Southern blot analyses can occur due to limited discrimination between 2 and 3 RCCX copies per genome [42].
2. PCR-based sequence analysis
Sanger sequencing allows the detection of point mutations and small insertions/deletions. PCR with oligonucleotide-specific primers designed for differential amplification of CYP21A2 (3.48 kb) and CYP21A1P (3.49 kb) (Fig. 5B) has been widely used. 31,43) The alleles containing the fusion gene can be detected using a primer set containing CYP21A1P-specific forward and CYP21A2-specific reverse primers. A CYP21A2-specific forward primer and a CYP21A1P-specific reverse primer can be used to detect alleles containing a rearranged gene. PCR products using CYP21A2-specific primer sets are analyzed by automatic direct sequencing to detect point mutations.
When designing primers for PCR-based sequencing, it is crucial to ensure that they are specific for the target gene or region of interest to prevent amplification of the pseudogene. Nonamplification by PCR can result from allelic dropout, which must be considered, as for any other gene [44]. Several single nucleotide polymorphisms in the promoters of both genes can interfere with primer binding, resulting in allelic dropout.
Another well-established technique is long-range PCR for an 8.5-kb fragment using CYP779f/Tena32F primers (Fig. 5C) that encompass the entire CYP21A2 gene and part of TNXB [45]. CYP779f (5'-AGGTGGGCTGTTTTCCTTTCA-3') is a common forward primer for the 5'-untranslated region (UTR) of the CYP21A2 and CYP21A1P genes. The CYP779f primer anneals from c.-799 to c.-779 based on the nucleotide sequences first described by Higashi et al. in 1986 (c.-802 to c.-782 based on NM_000500.9) [3,46]. The reverse primer (Tena32F) (5'-CTGTGCCTGGCTATAGCAAGC-3') anneals specifically to exon 32 of TNXB. This method can be used to detect TNXA/TNXB chimeras in patients with CAH-X syndrome. The amplified PCR products are then subjected to Sanger sequencing.
3. Multiplex-ligation dependent probe amplification analyses
The MLPA analysis is a robust method that can detect gene deletions, rearrangements, and fusion genes, and it requires only small amounts of DNA. A commercially available CYP21A2-MLPA kit (SALSA MLPA Probemix P050 CAH, MRC Holland, Amsterdam, The Netherlands) is widely used [44]. It contains 4 probes for the CYP21A1P gene and 8 probes for the CYP21A2 gene, enabling it to detect gene deletions, large gene conversions, a single nucleotide variant in the 5'-UTR (c.-113G>A), an 8 bp deletion in exon 3, p.I173N in exon 4, E6 cluster mutations in exon 6, and p.L308Ffs in exon 7. In addition, the probe mix contains 6 probes for the TNXB gene and can detect a 120 bp deletion at the boundary of exon 35 and intron 35 (CAH-X CH1). This method is limited to detecting variants that lie outside the target probe. Despite the challenges associated with genetic testing for 21-OHD, accurate genetic testing (up to 98%) can be achieved using PCR-based sequencing and MLPA [44].
4. Molecular genetic diagnosis of CAH-X syndrome
PCR-based detection of chimeric genes and MLPA analyses have been carried out to confirm the diagnosis of CAH-X syndrome [35]. PCR-based detection of chimeric genes has been performed using a mixed-primer strategy: a common forward primer for intron 43 of TNXA and TNXB and a reverse primer for intron 31 of TNXB (Fig. 5D). This produces a 5,010 bp fragment of TNXB, from part of intron 31 to part of intron 43 (Fig. 5D). This PCR product is used as a template for the subsequent sequencing analysis to assess TNXA/TNXB chimeras. CAH-X CH1 alleles are assessed by Sanger sequencing of exon 35 in TNXB, whereas CH2 and CH3 alleles are identified by direct sequencing of exons 40, 41, and 43. An MLPA analysis can detect a 120-bp deletion in TNXB exon 35 that is derived from the TNXA pseudogene. However, that is a laborious process and is not routinely performed. Therefore, the diagnosis of CAH-X syndrome relies primarily on clinical assessment using the Beighton 9-point scale [47].
5. Future directions
Recently, next-generation sequencing techniques have been increasingly used in clinical diagnostics, replacing traditional sequencing technologies. Because pseudogenes have high sequence homology, short read sequencing is limited in its ability to accurately map the reads to the correct genomic location. In addition, the complex genomic structure of RCCX module produces structural variations that are difficult to characterize using short-read sequencing technologies [48].
Therefore, long-read sequencing (LRS) technologies have been developed, such as single molecule real time (SMRT) sequencing (Pacific Biosciences, Menlo Park, CA, USA) and Oxford Nanopore sequencing (Oxford Nanopore Technologies, Oxford, UK) [49]. These techniques generate reads long enough to investigate repetitive or homologous regions. Long-range PCR encompassing the full-length of the CYP21A1P/TNXA and CYP21A2/partial TNXB genes that is followed by SMRT sequencing can detect various mutations, including point mutations and small insertions/deletions [50]. Locus-specific PCR using CYP779f/Tena32F followed by SMRT sequencing using the Sequel II platform (Pacific Biosciences) successfully identified pathogenic variants [51]. However, those studies showed that current LRS by PCR of a partial region is still limited in its ability to find CNVs and complex rearrangements. In addition, specialized bioinformatic algorithms are required to accurately detect mutations in highly homologous regions. Advances in bioinformatic tools and reductions in the cost of LRS-based testing will mitigate current challenges in the future.
Conclusions
The CYP21A2 gene is located on the RCCX module, which is a complex, multiallelic, and tandem CNV. This highly homologous gene structure can lead to genetic rearrangement during meiosis. Genetic testing is challenging due to the presence of pseudogenes and complex genomic structures. Recent extensive studies of the RCCX modules have led to a better understanding of the spectrum of TNX-related disorders. Identification of CAH-X syndrome is important to enable early intervention for musculoskeletal and cardiac abnormalities. The combination of clinical and biochemical markers with appropriate genetic testing promotes better outcomes in patients with 21-OHD.