Highlights
· Turner syndrome is caused by a partial or complete deletion of one of the X chromosomes. Xp deletion is a rarely reported cause of Turner syndrome. Here, we report the case of a 33-month-old girl who was diagnosed with Xp22.33 deletion combined with 7p22.3 duplication through chromosomal microarray.
To the editor,
Turner syndrome (TS) is a relatively common chromosomal disorder that affects one out of 2,000 female births. Clinical symptoms include short stature, cardiovascular and renal abnormalities, gonadal dysgenesis, and ovarian failure [1]. This syndrome is caused by partial or complete deletion of one of the X chromosomes. The degree of reproductive dysfunction can vary according to the range of deletion of the X chromosome. An Xp deletion is a rare cause of TS (2%). An entire Xp deletion can be linked to gonadal failure, whereas a terminal Xp deletion generally preserves reproductive functions [2-4]. Here, we report a 33-month-old girl who was admitted to the hospital with developmental delay and later diagnosed with Xp22.33 deletion combined with 7p22.3 duplication through chromosomal microarray (CMA).
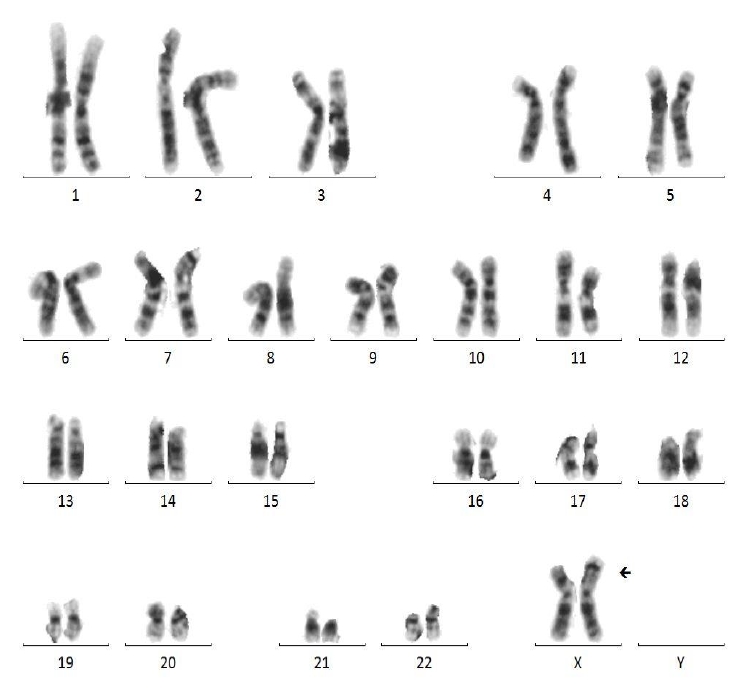
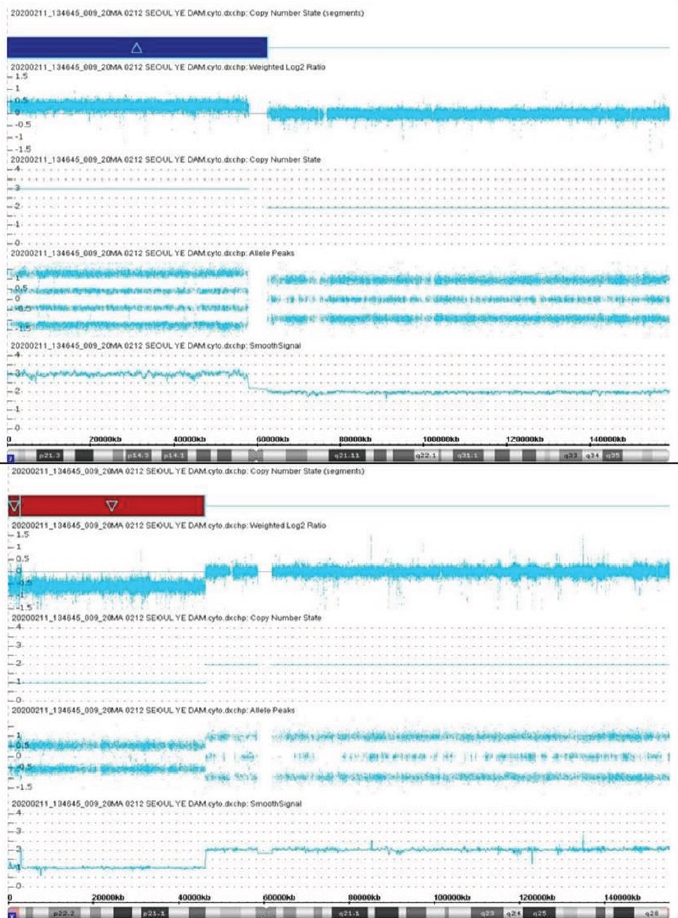
A 33-month-old girl was admitted due to a developmental delay with a short stature and Müllerian agenesis. She was born at 39 weeks of gestation from healthy, nonconsanguineous Korean parents, weighing 3,580 g (75th–90th percentile). The father's height was 179 cm (75th–85th percentile), and the mother's height was 167 cm (85th–90th percentile). On physical examination, the patient was relatively short-statured at 86.1 cm (3rd–5th percentile) and weighed 13.7 kg (50th–75th percentile), with a simian line present on both palms and joint laxity. We performed chromosomal and genetic analyses, and the results confirmed a karyotype of 46, X, der(X)t(X;7)(p11.23;p11.2) (Fig. 1). CMA confirmed Xp22.33-p11.3 deletion and 7p22.3-q11.21 duplication; 7p22.3q11.21(43,360–62,437,390) × 3, Xp22.33p11.3(168,546–46,178,795) × 1 (Fig. 2). The number of major genes involved in this site was 27 at the Xp22.33p11.3 site, including the short-stature-homeobox (SHOX) gene, and 13 at the 7p22.3q11.21 site. The symptoms that may appear due to this gene abnormality are similar to those of the reported patient: short stature, Müllerian agenesis of TS features, and developmental and skeletal anomalies of 7p duplication syndrome features. Whole-exome sequencing (WES) did not reveal any variants associated with the phenotypes. Thus, our patient was diagnosed with TS due to an Xp22.33 deletion combined with 7p22.3 duplication.
The 45,XO TS features characteristic clinical manifestations and is easily diagnosed by karyotyping. Deletion of any part of the X chromosome can result in gonadal failure, but a previous study reported that fertility was preserved even after loss of more than two-thirds of the short arm [4]. In addition, D'Ambrosio et al. [2] reported a case of Xp22.33-p22.12 deletion with preserved gonadal function, and Cho et al. [3] reported a case of familial TS (Xp22.33-Xp22.12) with preserved fertility. However, our case involved a relatively large deletion (Xp22.33-p11.3 deletion) and Müllerian agenesis. Furthermore, TS with atypical Müllerian agenesis (gonadal and Müllerian duct agenesis with a 46,X,i(Xq) karyotype) was previously reported by De Leon et al. [5] in 1984, while Vaddadi et al.[6] reported a case of Müllerian agenesis with a 45,X karyotype in 2013. However, a previous report of a patient with TS described absence of uterus and ovaries on pelvic ultrasound but later appearance on magnetic resonance imaging after estrogen replacement therapy, and the patient eventually developed regular menstrual cycles. Therefore, Müllerian agenesis must be diagnosed with caution in patients who do not undergo exploratory laparoscopy [7]. Our patient also had a dual diagnosis of 7p duplication that was linked to clinical symptoms, such as developmental delay, intellectual disability, behavioral problems, and distinctive facial features. Thus, the patient's Müllerian agenesis might have been caused by an Xp deletion [8].
Existing diagnostic tests for TS include karyotyping. However, CMA has a comparable level of accuracy to karyotyping and can better identify breakpoints in the presence of partial deletions or translocations [9]. Here, we performed CMA and WES in addition to karyotyping to determine the presence of various syndromes accompanying the developmental delay and Müllerian agenesis, but abnormal findings were only observed in karyotyping and CMA. WES generally sequences protein-coding regions and identifies single-nucleotide sequences and genetic variants; however, it does not include noncoding intron regions and is ineffective in identifying structural variations [10]. The patient showed clinical symptoms not caused by a single-nucleotide sequence mutation but instead by Xp22.33-p11.3 deletion and 7p22.33-q11.21 duplication. Therefore, the patient's condition could be diagnosed with karyotyping and CMA but not WES. As such, it is important to be aware of the features of each analysis and to choose the optimal test based on the patient's clinical symptoms or anticipated diagnosis.
To the best of our knowledge, this is the first report of TS due to Xp22.33 deletion combined with 7p22.3 duplication. We believe that our data will serve as a reference for analyzing atypical TS or Xp-deletion phenotypes.