Highlights
· The thyroid gland is a common target of autoimmune dysregulation.
· It is a highly responsive organ, capable of reacting to variable dietary iodine intakes.
· Balanced calorie intake is one of the most effective dietary strategies for affecting the thyroid cycle.
Introduction
The thyroid gland is a common target of autoimmune dysregulation [1]. In 1912, Hakaru Hashimoto, a Japanese physician, reported a syndrome in which lymphocytes invaded the thyroid, resulting in the production of antithyroid antibodies. Genetic, environmental, and dietary factors contribute to autoimmune thyroid disease (AITD), also known as autoimmune thyroiditis. AITD encompasses a number of thyroid gland disorders, ranging from hypothyroidism to hyperthyroidism. Hashimoto thyroiditis (HT) may cause hypothyroidism, while the most frequent causes of hyperthyroidism include Graves' disease [2]. Thyroid peroxidase (TPO) is the enzyme responsible for oxidizing iodide into iodine atoms for iodination of tyrosine residues on thyroglobulin (Tg), and subsequent oxidative coupling to produce the thyroid hormones (THs) monoiodotyrosine (T3) and diiodotyrosine (T4) from Tg. In HT, anti-TPO autoantibodies attack particular areas of the thyroid gland [3], leading to progressive thyroid damage, together with fatigue, weight gain, and constipation, reduced resistance to cold, dry skin, depression, muscular pains, and impaired exercise tolerance [4]. The prevalence of HT among women 60 years of age or older is 15%, and 2% among men in the same age group [5]. The primary autoantigen in Grave's disease (GD) is the thyroid-stimulating hormone (TSH) receptor, the targeting of which by anti-TSH antibody causes excessive TH production and symptoms that include agitation, fast heartbeat, loss of weight, poor heat tolerance, and bulging eyes (Graves' orbitopathy) [6]. Hypothyroidism may lead to weight gain secondary to edema, while substantial weight loss through enhanced muscle and adipose tissue catabolism may occur in hyperthyroidism. In addition to autoimmune dysregulation, the micronutrients iodine, iron, and selenium are nutritional variables that may alter thyroid function [7].
It has been estimated that there are approximately 0.8 cases of autoimmune-mediated hypothyroidism per 100 people, 95% of which are females. Worldwide, 2%–4% of women and 1% of men suffer from this disease, with the prevalence of the disease increasing with age [8]. The typical dietary iodide consumption in modern times frequently exceeds the recommended limit. Although a moderately high iodine intake through food is usually safe, THs must be synthesized at optimal levels. Excessive iodine intake is a risk factor for thyroid diseases, including AITD, and certain people are more susceptible, including the elderly, pregnant women, fetuses, and neonates, as well as those with pre-existing goiters or iodine deficiency. Either a deficit or excess of iodine may increase the incidence of thyroid disease, and iodine intakes should be maintained within a limited optimal range [9]. A daily intake of ~150–300 g of iodine is recommended by the World Health Organization (WHO; corresponding to a median urine iodine concentration of 100–199 g/L).
Dietary sources of iodine
Many foods and beverages lack iodine, a nutrient found in large quantities in the diet. Iodine concentration in drinking water varies by region, and is determined by soil iodine concentrations, ocean proximity, agricultural runoff, and water source and treatment technologies. As such, some countries, such as Israel, have low iodine concentrations in drinking water because desalinated water is used [10-12]. Iodine is found in vegetables and fruits, and the concentrations are mainly determined by soil iodine levels and the chemicals used in irrigation and fertilization. Iodine concentrations in plants cultivated on iodine-deficient soils will be higher than those grown on iodine-sufficient soils and vary between 10 μg per kg and 1 mg per kg. Dietary iodine intake of ovine animals, poultry, and beef cattle will be affected, but can be supplemented by feed and salt licks containing iodine-rich supplements [13]. There are varying amounts of iodine in hen eggs, ranging from 23 μg to 43 μg per 100 g [14]. Breast milk is rich in iodine, with concentrations 20–50 times greater than in plasma. In dairy products, iodine concentrations typically range from 33 to 534 μg/L (13 to 64% of the recommended daily consumption). Iodophors, which are iodine-based disinfectants used in milk production to clean udders and milk containers, may also contribute to iodine in dairy products [15]. Iodine is the most abundant element found in fish and marine plants due to its presence in seawater. Depending to the species, iodine concentrations in marine fish may range between 18 and 1,210 μg/100 g. Iodine concentrations in freshwater fish are approximately 6 times lower than those of marine fish, but the ranges of values are likely to overlap. Depending on the species of macroalgae (seaweed), iodine concentrations range from 16 μg per gram to over 8,165 μg per gram [16]. Seaweed use, formerly exclusive to Asian nations, has recently penetrated the global food industry, providing Western people with an additional source of iodine intake [17]. Seaweed that is high in iodine can, however, be harmful to the thyroid if consumed in excessive quantities, particularly by vulnerable groups, such as pregnant women and those with thyroid-related autoimmune issues [18-20].
Iodine intake and AITD
The WHO and the United States Institute of Medicine recommend that individuals consume iodine in amounts appropriate for their age and demographic category. Several studies have assessed the safety and effectiveness of iodine, and the benefits of iodine greatly outweigh any concerns iodine may cause. The WHO and the International Council for the Control of Iodine Deficiency Disorders adopted a global framework to prevent iodine deficiency. The use of iodized salt has been found to reduce iodine deficiency disorders and alter the pattern of thyroid illness [21]. Nonetheless, one significant effect of excessive iodine on thyroid function is termed the Wolf-Chaikof effect or long-term hypothyroidism caused by excess consumption of iodine. In addition, thyrotoxicosis associated with elevated iodine levels can cause acute toxic damage [22]. There is a well-established relationship between population iodine intake and nonautoimmune thyroid disorders, though there is less clarity on iodine's role in AITDs [23]. Several hypotheses have been proposed to explain the association of excessive iodine consumption and thyroid autoimmunity. First, iodine may be directly toxic to thyroid tissue and influence immune effector cells. Free radical damage to thyroid tissue is widely acknowledged to be a factor in the onset or exacerbation of AITD [24]. Experimental studies in genetically predisposed animals have shown that Tg becomes more immunogenic when exposed to excess iodine.
Thyroid function in infants and children
The developing fetus receives maternal T4 across the placenta, particularly during the first trimester of pregnancy, as well as hormones and other factors that affect thyroid function. Many studies of the effects of THs on the developing fetus have been conducted using sheep and rats, and the type of placentation and maturation timeframes must be considered when evaluating these findings. The rat thyroid gland is much less mature at birth than the human, and significant maturation of the rat thyroid gland and the hypothalamic-pituitary-thyroid axis occurs between birth and the first 2 or 3 weeks of life.
Newborns and children exhibit significant increases in TH turnover compared to adults, and their T4, free T4, T3, and TSH concentrations gradually decline from peak values in the early post-natal period, while reverse T3 concentrations in the serum remain stable or increase slightly. During the first year of life, serum Tg concentrations decrease similarly, reaching adult levels by the sixth month. Neonates produce an estimated 5 to 6 mg of T4 per kilogram daily; as a child grows, T4 production gradually declines to around 2 to 3 μg/kg/day at ages 3 to 9. In contrast, adults synthesize approximately 1.5 μg of T4 per kg daily. It is therefore imperative to use age-specific normative values when assessing serum T3. T4 and TSH concentrations in young children.
A newborn’s thyroid gland weighs around 1 g, and each lobe is approximately the size of the terminal phalanx of a newborn's thumb. The thyroid gland grows slowly during the first few months of a baby's life, and reaches its mature size of about 15 to 20 g at the age of 15 [25].
TH synthesis and metabolism
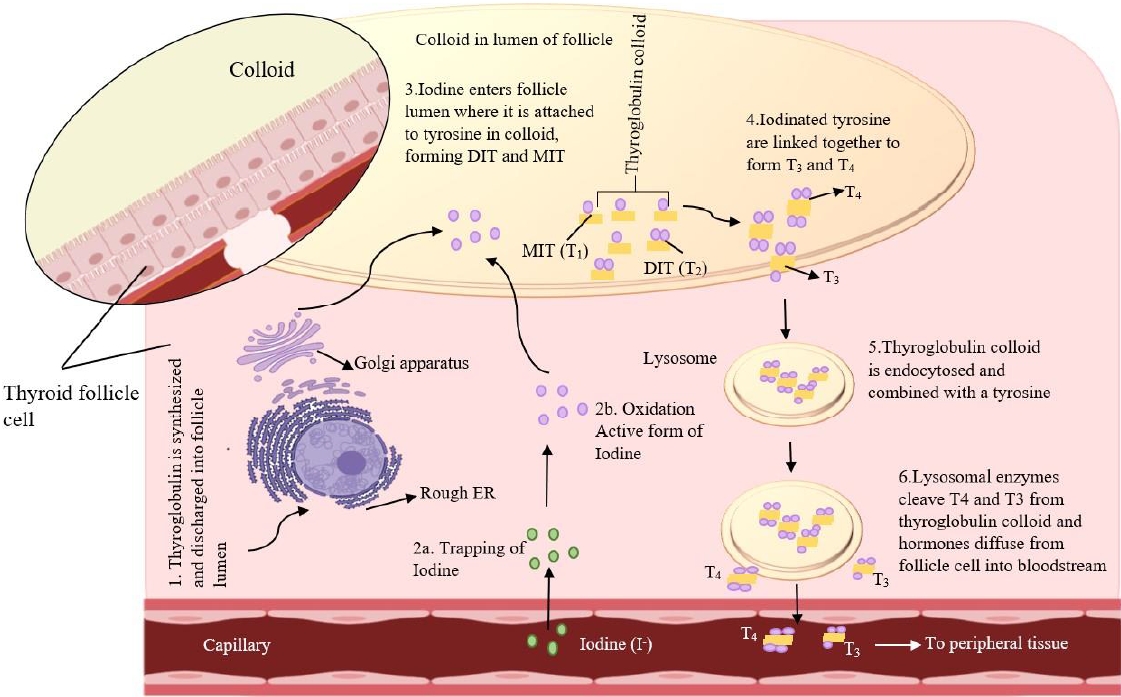
The thyroid gland produces 2 main hormones: thyroxine (T4) and triiodothyronine (T3). TSH is the primary regulator of TH synthesis and is present on the basolateral surface of thyrocytes. Thyrocytes receive iodine through sodium-iodine symporters (NIS) regulated by TSH; TSH also induces the creation of Tg, a glycoprotein produced by thyrocytes and stored within the follicular lumen (the colloid space). Iodide is combined with TPO, which oxidizes the iodide inside the cell to produce Tg. The Tg protease enzyme is activated by TSH to cleave Tg to generate T4, T3, and mono/T4 residues [26]. Hypothyroidism and goiter may result from a deficiency of any of the enzymes involved in TH synthesis. In addition, autosomal recessive mutations in the genes encoding these enzymes, often cause dyshormonogenic hypothyroidism [27] (Fig. 1).
The metabolism of iodine in the thyroid
Iodine stimulates the thyroid gland to produce THs [28]. The NIS symporter transfers iodide across the plasma membrane to reach the thyrocyte sodium concentration gradient established by the Na+/K+-ATPase transporter as the driving force [29]. Iodide is transported by multiple transporters into thyroid follicles, including PENDRIN, anoctamin-1, and cystic fibrosis transmembrane conductance regulators [30-32]. TPO oxidizing iodide to iodine radicals and then integrates them into particular tyrosine residues on Tg to form iodotyrosines. Two T4 residues combine to form T4 (tetraiodothyronine), while one T3 and one T4 residue combine to form T3. Lysosomal enzymes release the THs from the mature colloid of the follicular lumen through micropinocytosis and also remove degraded Tg protein from the lumen [33]. In the apical pole of thyrocytes, this enzyme uncouples protein residues by interacting with deiodinases, an enzyme involved in iodide recycling [34]. It transported THs from thyrocytes into the bloodstream via monocarboxylate transporter 8. Gene variants specific to thyroids that produce THs are activated by it [35].
Excessive iodine increases physiological responses
The thyroid gland is a highly responsive organ, capable of reacting to variable dietary iodine intakes and, when stimulated, of accumulating iodine at up to an 80-fold concentration gradient. Most euthyroid people can consume up to 2 g of iodine per day without experiencing a clinical response if they do not have underlying thyroiditis and live in iodine-sufficient areas. At high iodine intakes, only slight changes in TH concentrations are evident: serum T4 and T3 concentrations may decrease by 25% and 15%, respectively, with a corresponding rise in TSH of 12 mIU/L, even though these values remain within the reference interval in most people. Although ultrasonographic thyroid volume may mildly increase, goiter or thyroid dysfunction does not appear clinically. Most of these mild side effects are reversible [18].
Control of the thyroid by iodine
Iodide influences thyroid function, with its principal activities limiting the thyroid's reactivity to thyrotropin and initially avoid oxidation. At elevated levels, the release of THs is stifled so that its trapping is reduced after a delayed period. Modification of dietary iodine intakes may be used to adjust blood TSH levels. Iodide control of the thyroid response to TSH [36] comprises a negative feedback loop. Iodine organification (the incorporation of iodine into Tg) increases initially and then decreases as the iodide intake increases. The Wolff-Chaikoff effect is reliable for inorganic iodide concentration within the thyroid cells. Although the mechanism is unknown, iodide's inhibition of TPO or other enzymes may contribute to inhibition of iodine organification [37].
Environmental factors influence the pathogenesis of thyroid disease
Regions with higher soil and/or water iodine content tend to have a higher prevalence of autoimmune hypothyroidism and antithyroid antibodies. The most current evidence from Denmark supports this claim [38]: prior to iodization of salt, the prevalence of anti-TPO antibody (anti-TPO-Ab) was 14.3%, but increased to 23.8% after iodization; the prevalence of overt hypothyroidism increased from 38.3/100.000 per year at baseline to 47.2/100.000 per year a few years later. In a small Italian community, voluntary iodine prophylaxis increased the incidence of anti-TPO-Ab and hypothyroidism 15 years later. The World Health Organization recommends that everyone consume 150 g of iodine daily [39]. The risk of GD has historically been linked to smoking, with an almost 3-fold greater likelihood of developing Graves' hyperthyroidism and/or Graves’ orbitopathy among smokers. Symptoms typically disappear a few years after cessation of smoking, are dose-dependent, and are less pronounced in women. Smoking is also postulated to help prevent hypothyroidism, although there have been few studies of this effect. The prevalence of anti-TPO-Ab has been reported to be lower in smokers than in nonsmokers [40].
Alcohol has also been proven to offer protection against autoimmune diseases, including type-1 diabetes mellitus and rheumatoid arthritis. The preventive effects of alcohol are not yet known to have a specific mechanism of action [41]. Thioredoxin reductases and selenium-dependent glutathione peroxidases control cellular redox status and protect cells from oxidative damage. Compared with other organs, the thyroid gland contains the greatest amount of selenium per gram. Immune function can be impaired when selenium levels are low. Therefore, a minor selenium deficiency may exacerbate autoimmune thyroid illness. Supplements containing selenium have been reported to reduce postpartum thyroid dysfunction and anti-TPO-Ab levels during pregnancy [6,42]. Stress is widely acknowledged to play a role in the onset of GD. There is evidence that patients with Graves' hyperthyroidism experience more stressful life events prior to their diagnosis than controls; however, these investigations were all conducted retrospectively, and the evidence is only circumstantial. There has been minimal research into the role of stress in the development of HT [43]. It is challenging to apply thyroid-related toxicant findings to an individual since thyroid-related toxicant assessments examine the effects on the general population (Table 1) [44].
Apoptosis and AITD
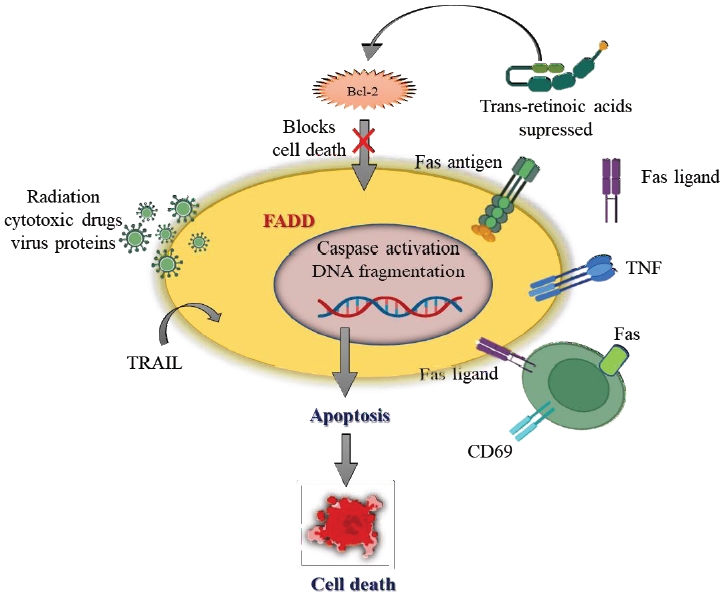
Even though both inherited and environmental factors contribute to the pathogenesis of AITD, it remains unclear how the disease develops. In HT, apoptosis is increased in situ, whereas in GD, apoptosis is reduced, underscoring that apoptosis regulates cell function. Invading lymphoid cells make up a large proportion of apoptotic cells within the thyroid. Invading CD4 T lymphocytes kill Fas-ligand (FasL)-expressing target cells in the thyroid glands in people with GD. Thyrocytestimulating antibodies that stimulate thyrocyte hyperplasia may be less cytotoxic on activated FasL-expressing CD4 T cells. Besides TSH, IgG from GD patients reduces Fas expression in thyrocytes. Thyroid-stimulating antibodies reduce Fas production by increasing cyclic adenosine monophosphate (cAMP) in thyroid cells. Moreover, anti-TSH receptor antibodies may suppress Fas-mediated apoptosis in GD patients, leading to the development of goiter. In addition, TSH inhibits cAMP production and Fas expression in thyrocytes of patients with idiopathic myxedema.
Lymphocytes become more efficient when incorporated self-antigens, and soluble Fas receptor competes with membrane-bound Fas receptor for lymphocyte proliferation and development. It is also important to note that soluble Fas suppresses the Fas and FasL systems in Graves' disease patients. Untreated individuals with the illness had significantly higher serum levels of soluble Fas than age-matched control participants. In patients with GD, soluble Fas levels decline after 6 to 8 weeks of antithyroid medications. As far as TSH, antimicrosomal antibodies, and antithyroglobulin antibodies are concerned, there is no correlation between soluble Fas levels and anti-TSH receptor antibodies. Interleukin 1α and tumor necrosis factor-β have been reported to promote the production of soluble Fas in the supernatant of cultured thyrocytes from GD patients. These conditions are associated with soluble Fas contributing to GD development (Fig. 2) [45].
Genetics plays a vital role in the pathogenesis of AITD
To decipher the complex genetic contribution to autoimmune diseases, certain types of DNA variants and methodologies accounting for gene-environment interaction, genomegenomic interaction, and transgenerational inheritance findings have been examined [46,47]. The susceptibility of loci clustering in the same immunological pathway or nearby GD may be affected by gene-gene interactions between human leukocyte antigen (HLA) category II molecules and Tg. It is believed that environmental factors account for about 20% of the predisposition to AITD in monozygotic twins [48]; the lack of concordance between monozygotic twins has highlighted this factor. Several genes implicated in regulating immune responses are found on the short arm of chromosome six, known as the HLA complex (a correlate of the major histocompatibility complex in laboratory animals). There are 3 histocompatibility classes: Class I contains genes encoding immune-responserelated molecules and molecules that are no longer associated with immunity; Class II comprises genes encoding immuneresponse-related molecules; and Class III includes genes encoding other molecules not associated with immunity [49]. In immunological checkpoints, CTLA-4 functions as a protein receptor called CD152 (cluster of differentiation 152). This protein controls the immune response at the surface of T cells. In addition to reducing mRNA synthesis, polymorphisms encoding the soluble form of the molecule are thought to be beneficial. Many It has linked gene variations to GD, the most likely a polymorphism in position 60 [50].
Significant genes play a role in AITD
Antithyroid antibodies have been linked to several genes implicated in AITD, as shown in Table 2. By utilizing conventional case-control studies and screening for single nucleotide polymorphisms, the following AITD-associated genes were identified: CD4+ T-helper cells recognize foreign antigens with HLA class II molecules, while the interleukin-2 receptor alpha chain encodes CD25, a protein suppressed by T-regulatory cells. In addition, by interacting with T-cell receptor signaling molecules, protein tyrosine phosphatase nonreceptor type 22 takes part in T-cell signal transduction; CTLA4 inhibits T-cell signaling, and HLA class II molecules inhibit exogenous antigen presentation. Furthermore, genome-wide association studies (GWAS) and case-control studies found additional AITD genes, including Fc-receptor-like 3, which plays both a positive and a negative role in B-cell signaling. In addition, endogenous antigens, such as virally derived antigens, are presented to CD8+ T cells by HLA class I molecules; TSHR is the primary autoantigen target in GD and is the TSH receptor.
The GWAS and genotyping microarray (e.g., Immunochip) results identified the following AITD-associated genes: CD4+ T-helper cells and CD8+ T cells express GD candidate gene at 4p14 (GDCG4p14), and CD4+ T-helper and CD8+ T cells express RNASET2. BACH2 may influence B-cell development and antibody production through its role in B-cell maturation. Tg and TPO promoters contain forkhead box E1 (FOXE1) response elements that aid thyroid gland morphogenesis. There is no clear understanding of the function of other AITDassociated genes identified by GWAS and Immunochip. The immunopathogenesis of AITD may therefore be influenced by susceptibility genes with known functions, indicating the relevance T cells to the pathogenesis of AITD [51].
The GDCG4p14 is a newly discovered gene whose 5' ends are located approximately 5 kb downstream of the shorter cDNA [52]. The small Maf family gene BACH2 can switch between repressor and activator functions. It is significant to note that BACH2 is expressed at 4 stages in primary B cells: pro-, pre- , immature, and mature. A role for BACH2 may be played by primary B cells in developing and producing antibodies [53]. In humans, only RNASET2 is a member of the RNase T2 family, and it is thought to play a role in inhibiting tumorigenesis, metastasis, and angiogenesis. In human dendritic cells, the RNase T2 family secreted from Schistosoma mansoni eggs primes CD4+ T cells for Th2 polarization. According to current theories, Graves' disease is caused by an abnormal immune response triggered by RNASET2 [52]. A thyroid transcription factor, the thyroid transcription factor 2, is also known as the FOXE1 and was previously known as forkhead drosophila homolog-like 15. This transcription factor belongs to the family of transcription factors with a forkhead/winged helix design. Thyroid-specific proteins recognize and bind to DNA sequences in the promoters of the Tg and thyroperoxidase (TPO) genes specific to thyroid follicles. As an adult thyroid gland transcriptional repressor, this protein may also suppress the expression of both thyroid genes [54].
Oxidative stress in TH physiology
Reactive oxygen species (ROS) may contribute to the underlying cause of many illnesses because of their critical role in many physiological processes. Hydrogen peroxide (H2O2) is a diffusible second messenger involved in redox processes that acts as a signaling molecule [55]. While oxidative processes occur in all tissues and organs, ROS play a specific role in thyroid physiology. THs are produced. Iodine is the only external element, along with TPO, H2O2, and Tg [56,57]. The enzyme TPO oxidizes iodide to iodine with H2O2 acting to accept electrons from the reaction between iodine and iodotyrosine. Oxidative processes are at the basis of both the Wolff-Chaikoff effect and escape phenomenon. Under physiological conditions, the thyroid's endogenous antioxidative system is balanced against ROS formation, which is required for TH production. Processes causing imbalances in these various thyroid conditions can cause oxidative damage to macromolecules [58].
Conclusions
Nutrition strongly influences the supply, regulation, and disposal of the THs that play a crucial role in controlling the body's metabolism. Therefore, dietary strategies to influence the thyroid cycle must comprise a balanced intake of calories with a proper iodine supply. Caloric restriction alone most likely leads to reductions in catabolic expenditure due to homeostatic controls. When iodine supply to the thyroid gland in inadequate, compensatory alterations in iodide accumulation and binding occur in the thyroid gland to ensure appropriate synthesis and supply of THs. Despite widespread iodine supplementation in many Western countries, goiter and other illnesses may resurface due to insufficient monitoring of iodine intakes, vegetarianism, and reduced salt consumption. In addition, iodine intake can exacerbate thyroiditis in amiodarone users by increasing Tg autoantibodies. Iodine supplementation increases the frequency and severity of autoimmune thyroiditis in animal models, which is consistent with epidemiological studies. Iodization of salt remains the most successful strategy to eradicate iodine deficiency in human diets. However, the hazards (and iodineinduced hyperthyroidism, in particular) exceed the advantages. Therefore, doctors and patients need to be aware of the potential to induce thyroid abnormalities as a result of the use of specific meals and herbal medicines.