Highlights
· Genetic obesities should be suspected in children with early-onset morbid obesity and hyperphagia. Variants in single gene involved in the leptin-melanocortin pathway cause monogenic obesity. Early genetic evaluation enables to identify treatable obesity and provide personalized management.
Introduction
According to endocrine society practice guideline for pediatric obesity, the childhood obesity is defined as body mass index (BMI) ≥ 95th percentile, and extreme obesity as BMI ≥120% of the 95th percentile or ≥35 kg/m2 [1]. The prevalence of extreme childhood obesity reported as ranges from 1.96% to 6.3%, depending on criteria, in the general population [2,3].
Childhood obesity leads to not only expansion of adipose tissue mass but also dysfunction of adipose tissue including adipocyte hypertrophy, inflammation, and dysregulation of adipokine secretion from early life [4]. The early-onset obesity is a serious health problem as it can cause cardiovascular and metabolic comorbidities, consequently related with increased mortality in adulthood [5]. Obese children are often suffered from stigmatization and associated psychological problems leading to restriction of social life. Therefore, early diagnosis and timely appropriate management are important in childhood obesity.
Based on the genetic contribution, childhood obesity can be classified into 3 groups. Majority of childhood obesity is classified as "common polygenic obesity" which resulted from interaction of environmental factors and genetic susceptibilities [3]. "Syndromic obesity" is often considered when the child has obesity with developmental delay or dysmorphic features suggesting specific genetic syndromes including Prader-Willi syndrome (PWS), Bardet-Biedle syndrome (BBS), and Alstrom syndrome. "Monogenic obesity" is caused by variants in single gene which are usually involved in the regulation of hunger and satiety associated with the hypothalamic leptin-melanocortin pathway in central nervous system [3].
If a patient has early-onset extreme obesity before 5 years of age, it would be a 'red flag sign' for genetic obesity and indicative subsequent genetic evaluation to identify underlying molecular genetic defect. As new precision medicine approaches based on genetic etiology are emerging, this review focuses on the genetic obesity including "syndromic obesity" and "monogenic obesity" associated with leptin-melanocortin pathway deficit.
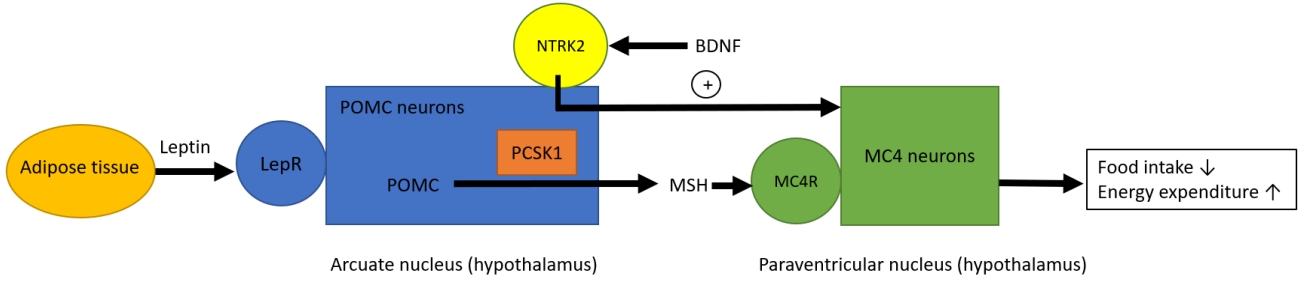
Leptin-melanocortin pathway
Many of genetic obesity disorders are resulted from defects in leptin-melanocortin pathway (Fig. 1). This neural pathway is responsible for regulating appetite, satiety, and body weight [6]. Leptin produced and secreted from adipose tissue acts at leptin receptor in the arcuate nucleus of the hypothalamus. It activates increased production of pro-opiomelanocortin (POMC) in POMC neuron, which is further processed to melanocytestimulating hormones (MSH) by proprotein convertase subtilisin/kexin type 1 (PCSK1) [6]. MSH acts centrally at the melanocortin receptors, including the melanocortin 4 receptor (MC4R). Activation of MC4R leads to reduction of food intake and increased energy expenditure [6]. Brain-derived neurotrophic factor modulates leptin-mediated synaptic plasticity of neurons [3].
Syndromic obesity
The clinical characteristics of syndromic obesity is early-onset obesity accompanied by other clinical manifestations including dysmorphic features, short stature, congenital anomalies, and neurodevelopmental deficits such as developmental delay, intellectual disability, and autism spectrum disorders [3,7]. More than 80 distinct syndromic obesities have been described to date.8) Most common syndromes including PWS, BBS, and Alstrome syndrome share pathophysiology related to hypothalamic impairment [3,7,8].
1. Prader-Willi syndrome
The frequency of PWS is between 1/15,000 and 1/20,000 births [8]. The clinical manifestations include neonatal hypotonia, hyperphagia starting from 2–3 years of age leading to obesity, and abnormal body composition with increased fat mass and reduced lean body mass [9]. Hypogonadotropic hypogonadism, growth hormone deficiency, intellectual disability, learning difficulties, behavioral problems might be accompanied. The patients with PWS usually develop severe obesity related to uncontrolled eating behavior during evolution from childhood to adulthood [10]. PWS is caused by abnormal parental genomic imprinting with functional absence of paternal chromosomal segment 15q11-q13 [10]. Management of PWS requires a multidisciplinary team approach including neonatologist, medical geneticist, pediatric endocrinologist, dietitian, psychologist, orthopedist, speech therapist, and physical therapist [11-13]. Despite several clinical trials for pharmacologic treatment of obesity in PWS have been performed including glucagon-like peptide 1 agonist, oxytocin/carbetocin, MC4R agonist, no drug yet has shown consistent beneficial effect to date [12].
2. Bardet-Biedle syndrome
BBS is an autosomal recessive genetic disorder presented with heterogeneous clinical symptoms [8]. Patients with BBS may have retinal dystrophy, polydactyly, kidney abnormalities, hypogonadism, and learning difficulties in addition to earlyonset obesity. BBS is genetically heterogeneous ciliopathy and at least 19 genes related to function of the primary cilium are involved [14]. Although the exact mechanisms of obesity in BBS are still uncertain, central origin obesity due to hypothalamic dysfunction associated with hyperphagia and peripheral origin involving proliferation of adipose tissue or other endocrine tissues such as pancreas, intestines have been propsed [8,15,16]. Treatment of obesity in BBS using MC4R agonist was evaluated and showed beneficial effect in clinical trials [3,17,18].
3. Alstrom syndrome
Alstrom syndrome is characterized by obesity accompanied with cone-rod dystrophy, progressive bilateral sensorineural hearing loss, cardiomyopathy, insulin resistance, and chronic progressive kidney disease [19]. Majority of the patients experience progressive visual impairment leading to loss of all light perception by their second decade. Affected children usually have normal birth weight but develop truncal obesity during the first year of life. Approximately 20% of patients have delayed developmental milestones [19]. However intellectual disability is rare despite 30% of affected individuals have learning disability [19]. The molecular diagnosis can be established by identification of biallelic pathogenic variants in ALMS1 [20]. A phase III clinical trial for evaluating efficacy and safety of MC4R agonist in patients with Alstrom syndrome was performed and showed beneficial effect [18].
4. Albright hereditary osteodystrophy
Albright hereditary osteodystrophy (AHO) is resulted from a pathogenic variant in GNAS which encodes Gαs mediating G protein-coupled receptor signaling [3]. Clinical symptoms of AHO include early-onset obesity, short stature, round facies, shortening of 4th and/or 5th metacarpal and metatarsal bones, and subcutaneous ossification [21]. As GNAS is one of the imprinted genes, pathogenic variants in maternal allele cause not only AHO but also hormone resistances including parathyroid hormone (pseudohypoparathyroidism), thyroid-stimulating hormone, gonadotropins, and brain neurotransmitters [21]. Nevertheless nearly all pathogenic variants in GNAS lead to impairment of MC4R signaling [3,22], no specific pharmacologic treatment for obesity in patients with GNAS pathogenic variants has been available yet
Monogenic obesity
BMI trajectories of children with monogenic obesity are clearly different from those of common polygenic obesity. Children with monogenic obesity generally shows rapid weight gain started early after birth, with the fastest increase of BMI in the first year of life [3]. Central hypothalamic and neuroendocrine pathways are often impaired in monogenic obesity [23]. However, monogenic obesity could be differentiated from syndromic obesity by the fact that cognitive development is usually normal in children with monogenic obesity [3]. Monogenic obesity should be suspected if patients have severe obesity (BMI≥120% of the 95th percentile or ≥35 kg/m2) before 5 years of age, rapid weight gain in the first 2 years of life, hyperphagia, additional clinical manifestations to obesity including short stature, red hair, adrenal insufficiency, hypothyroidism, hypogonadism, pituitary insufficiencies, diabetes insipidus, increased predisposition to infection or intractable recurrent diarrhea [3].
1. MC4R deficiency
Loss of function mutations in MC4R account for 2%–5% of severe early-onset obesity [6,24] and MC4R deficiency is the most frequent form of monogenic obesity to date [3,23]. The mutations in MC4R can be inherited either in a dominant or recessive pattern. Clinical manifestations in recessive forms are more severe than those of dominant form. Patients with MC4R deficiency have hyperphagia and extreme early-onset obesity, severe hyperinsulinemia along with increased lean body mass and accelerated linear growth [1]. In contrast, patients with gain of function mutations in MC4R is known to have reduced food intake, lower BMI, and increased energy expenditure [25].
2. Leptin deficiency
Leptin deficiency results from the pathogenic variants in leptin gene (LEP). The variants can be symptomatic in either heterozygous or homozygous forms [26]. The clinical symptoms of heterozygous state include severe hyperphagia, early-onset obesity, while those of homozygous state can have additional symptoms such as frequent infections, hypogonadotropic hypogonadism and hypothyroidism [27]. The circulating leptin levels are nearly nondetectable in patients with leptin deficiency [28].
3. Leptin receptor deficiency
Leptin receptor deficiency is a rare autosomal recessive disorder caused by homozygous or compound heterozygous pathogenic variants in LEPR [29]. The phenotype is largely shared with leptin deficiency except increased circulating levels of leptin [29]. Patients with leptin receptor deficiency usually have normal birth weight, but exhibit prominent foodseeking behavior, hyperphagia, and impaired satiety leading to rapid weight gain and severe obesity [30]. Accompanied hypogonadotropic hypogonadism and pituitary insufficiency are comparable symptoms of leptin deficiency, whereas recurrent infections are less frequent [29,30].
4. POMC deficiency
POMC deficiency is an autosomal recessive disorder characterized by hyperphagia leading to severe early-onset obesity, adrenal insufficiency, and pigmentary abnormalities including pale skin or red hair. POMC encodes a pituitary preproprotein that is a precursor of several neuroendocrine peptides including α-MSH, which is processed by the PCSK1 and acts via the MC4R to suppress appetite and food intake [31].
5. PCSK1 deficiency
PCSK1 deficiency present with complex clinical symptoms including severe malabsorptive diarrhea in the neonatal period, postprandial hypoglycemia and early-onset obesity [32,33]. Patients may also have neuroendocrine problems including hypogonadotropic hypogonadism, diabetes insipidus, hypothyroidism, and adrenal insufficiency [32,33]. Biallelic mutations in PCSK1 cause impaired processing of prohormones to active hormones. gastric peptides and proinsulin [3,32,33].
6. SH2B1 deficiency
The SH2B adapter protein 1 (SH2B1) is a key molecule in leptin-mediated signal pathway that activates the downstream signal by JNK2-dependent and JNK2-independent mechanisms [34]. SH2B1 also serves as adaptor molecule in the insulin signal cascade and act as a regulator of glucose homeostasis and insulin sensitivity [35]. SH2B1 deficiency is a autosomal recessively inherited monogenic obesity [3]. Patients with SH2B1 deficiency have hyperphagia, early-onset obesity, severe insulin resistance along with maladaptive behaviors [36].
Diagnostic strategies
Genetic evaluations such as gene panel testing or exomebased sequencing is highly recommended in patients with earlyonset severe obesity before the age of 5 years, along with hyperphagia and/or family history. [1,6]. If there is coexisting features of syndromic cause including developmental delay, dysmorphic features, hormonal deficit, or vision loss, genetic tests targeting the individual syndromic obesity should be considered [6]. Early genetic diagnosis is necessary in severe early-onset obesity to avoid the frustrating failure of general life-style modification for common obesity [3]. Dayton and Miller [6] suggested diagnostic algorithm for early diagnosis of genetic obesity (Fig. 2). Genetic and clinical implications of syndromic or monogenic obesities are summarized in Table 1.
Emerging pharmacological treatments
In recent years, genetic analysis has enabled development of new pharmacological therapeutic approached for several types of genetic obesity. The development of therapies for a genetic obesity is crucial as it is highly probable that a drug developed for one disorder may be effective for wide spectrum of genetic obesities because these genetic disorders involve common biological pathways [8]. Moreover, common polygenic obesity might also benefit from these therapeutic approaches in the future given that genetic trait of obesity also play a role even in common obesity.
1. Metreleptin
Congenital leptin deficiency or leptin dysfunction can be treated by hormone supplementary therapy. Subcutaneous administration of human recombinant leptin at 0.03 mg/kg of lean body mass daily dose leads to rapid amelioration of eating behavior, a reduction of food intake, and subsequent loss of body weight [37]. It was also effective in improvement of metabolic and endocrine dysfunctions including hyperinsulinemia, hyperlipidemia, and liver steatosis as well as central hypogonadism [3,37]. Adverse effects of metreleptin include the formation of neutralizing anti-leptin antibodies, and an increased risk of lymphoma [3,38].
2. Setmelanotide
Setmelanotide is a MC4R agonist introduced in 2016 [39]. MC4R agonist acts by mimicking the POMC derivative α-MSH that could result in reducing food intake and substantial weight loss. Setmelanotide showed beneficial effects in 2 patients with POMC deficiency in a phase II clinical trial [39]. Given that the most genes implicated in monogenic obesity are involved in MC4R pathway, setmelanotide has been additionally investigated in patients with defects in the central leptinmelanocortin pathway: leptin receptor deficiency and MC4R deficiency. In a phase III clinical trial, 80% of the patients with POMC deficiency and 45% of 11 patients with LEPR deficiency lost at least 10% of body weight after 1 year [40]. In 2020, setmelanotide got U.S. Food and Drug Administration approval for management of obesity in adult and children aged 6 years and older with monogenic obesity due to POMC, PCSK1, or leptin receptor deficiency.41) Setmelanotide is currently investigated in phase II or phase III clinical trials as a treatment option of many other genetic deficits in the MC4R pathway [3]. In addition, setmelanotide is also being evaluated in patients with syndromic obesity such as BBS and Alstrom syndrome as well as chromosomal rearrangement of the 16p111.2 locus [3,17,18].
3. Oxytocin
Oxytocin impact on the downstream of the leptin-melanocortin pathway. Administration of oxytocin enhances the activity of brain regions that exert cognitive control, while concomitantly increase the activity of structures that process food-reward value with overall reduction of food intake [42]. Peripheral administration of oxytocin prevents the decrease in metabolic rate accompanies weight loss, likely by inducing lipolysis and fat oxidation [43].
Innovative therapeutic approaches
Beyond the novel medications for genetic obesity, there are other innovative approaches for obesity and appetite regulation. Patients with monogenic obesity may have beneficial effect by applying novel induced pluripotent stem cell (iPSC) and CRISPR-Cas9 mediated gene editing technologies [44]. Despite iPSC therapy had enormous clinical potential, limitations including tumorigenicity and immunogenicity are still existing for clinical application [45]. CRISPR-Cas9 mediated in vivo or ex vivo gene editing aim to functionally repair defective genetic variants causing monogenic obesity [46]. However, comprehensive preclinical and clinical studies are necessary to apply these novel gene therapies to clinical practice.
Conclusions
Once obesity has been established in children and adolescents, it has a high risk of persisting in to adulthood and leading to comorbidities. Early genetic evaluation enables to identify treatable obesity and provide timely intervention which may eventually achieve favorable outcome by establishing more personalized management.