Highlights
· We report a case of new GNRHR compound variant c.514G>A (p Gly172Arg) and c.113G>A (p.Arg38Gln) in a Korean adolescent admitted to our hospital for hypogonadotropic hypogonadism.
To the Editor,
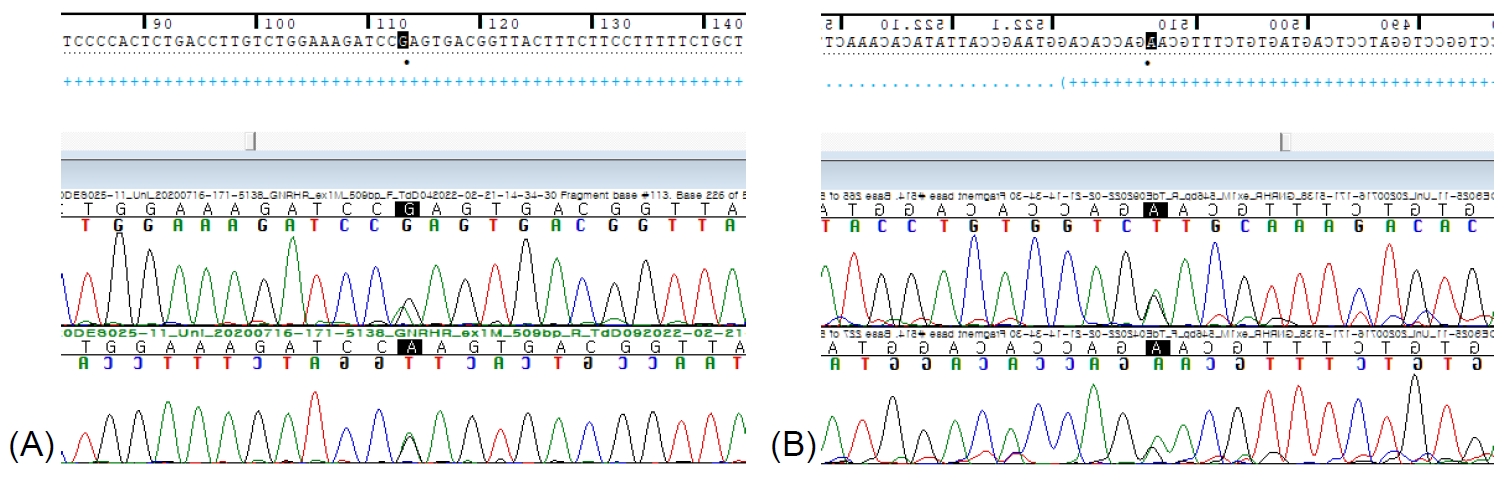
Congenital idiopathic hypogonadotropic hypogonadism (IHH) is a disorder characterized by incomplete or absence of sexual maturation by the age of 18 years. Aside from low serum circulating gonadotropin and testosterone levels, patients with IHH show no other abnormalities of the hypothalamic–pituitary–gonadal axis [1]. Mutation of the gonadotropin-releasing hormone receptor gene (GNRHR) is reported to be the causal factor in 16% of IHH cases [2]. Hypogonadotropic hypogonadism 7 (HH7), with or without anosmia, is a subtype of IHH caused by a homozygous or compound heterozygous mutation in GNRHR, located on chromosome 4q13. Although the phenotypes of patients with HH7 vary, partial or complete IHH is common depending on the level of abnormality in the hypothalamic–pituitary–gonadal axis [3]. GNRHR belongs to the superfamily of G-protein–coupled receptors, and its mutation causes abnormalities in the synthesis of receptors, trafficking and/or internalization to the cell membrane, recycling of receptors, ligand binding, and signal transduction [4]. Biallelic mutations in GNRHR, the gene expression of IHH by GNRHR mutation, have been reported several times, and they lead to hypogonadotropic hypogonadism with normosmia [5]. In Korea, no case of GNRHR mutation in an adolescent related to IHH has been reported. An 18-year-old male adolescent was referred to the pediatric endocrinology department of our hospital due to delayed puberty during treatment for nephrotic syndrome. Physical examination showed that the stretched penile length was 7 cm (normal reference, 13.3±1.6 cm), and he had a scrotum but no palpable testes [6]. The patient had exhibited cryptorchidism and small testis at birth and had undergone bilateral orchidopexy at 1 year of age. After orchidopexy, he was observed for progression of puberty during regular visits to the urology department, and he was transferred to the endocrinology department around the age of 7 years old. The karyotype was 46,XY. Genetic testing showed a negative result for Prader-Willi syndrome with methylation-specific polymerase chain reaction, and no mutation was found on the WT1 gene. At the age of 9 years, he visited the hospital with repeated proteinuria and edema, where he was diagnosed with steroid-resistant focal segmental glomerulosclerosis and treated with continuous steroid therapy and immune-suppressants. His progress was monitored without any further testing or treatment regarding pubertal progression. After a long absence of follow-up for hypogonadism, he was referred to us again at the age of 18 years because of delayed puberty. The hormone test results of the patient at 7 and 18 years old are summarized in Table 1. The SRY gene test was positive. Panel exome sequencing showed a compound heterozygous missense variant of c.514G>A (p.Gly172Arg) and c.113G>A (p.Arg38Gln) in the GNRHR gene (Fig. 1). After the mutation in the GNRHR gene was confirmed, the younger brother underwent a genetic test at another hospital, and a compound heterozygous variant in the same region of the GNRHR gene was identified. His parents refused to undergo genetic testing, so the mutation could not be confirmed in either of them. At present, we have confirmed that our patient’s testosterone level increased to ≥6.6 ng/mL, and he is undergoing continuous treatment with a 250-mg injection of testosterone at 4-week intervals. We report a newly identified case of complete IHH in a young male Korean patient caused by a compound heterozygous missense variant of c.514G>A (p.Gly172Arg) and c.113G>A (p.Arg38Gln) in the GNRHR gene, along with the hormone test results and progress of the patient.