Highlights
┬Ę Hypothalamic hamartomas (HHs) are noncancerous growths in the hypothalamus causing central precocious puberty and gelastic seizures. Seckel syndrome 5 (SCKL5) is a rare genetic disorder with growth issues, intellectual disability, unique facial features, microcephaly, and brain abnormalities. This report presents cases of 2 siblings had short stature and were diagnosed with SCKL5. Despite sharing the same genetic variant, their phenotypes differed, a previously unreported occurrence. This study also presents the first genetically confirmed cases of SCKL5 in Korea.
Introduction
Hypothalamic hamartomas (HHs) are congenital, nonprogressive, and nonneoplastic mass lesions located in the ventral hypothalamus. Patients with HH present with peculiar symptoms, including central precocious puberty (CPP) and/or gelastic seizures [1,2]. The major phenotype of patients with HHs that arise in the anterior hypothalamus is CPP [2]. Seckel syndrome 5 (OMIM613823) is a rare genetic disorder of intrauterine growth retardation that results in low birth weight. It is an autosomal recessive neurodevelopmental spectrum disorder characterized by a small head circumference; proportionate osteodysplastic primordial dwarfism; growth retardation; a wide spectrum of intellectual disability; and unique facial features including a beak-like nose, sloping forehead, narrow face, large eyes, and micrognathia [3,4]. Since it is a spectrum disorder, many causative genetic variants have been identified in centrosomal proteins linked to microcephaly and dwarfism [5]. Variants in the CEP152 gene, which is located on chromosome 15q21.1, have been identified as the cause of Seckel syndrome 5 (SCKL 5). The gene encodes a core protein of the centrosome that affects cell shape, polarity, and division. In previous reports, accompanying brain structural malformations and vasculopathies have been reported in patients with SCKL [6]. However, there have been no previous reports of SCKL with HH related to CPP. Here, we report a case of 2 siblings with the same genetic variant of CEP152 who presented with different phenotypes that had not been reported previously. They were also the first SCKL 5 patients diagnosed by genetic analysis in Korea.
Case reports
Two sisters were referred to Inha University Hospital for investigation of short stature.
1. Case 1
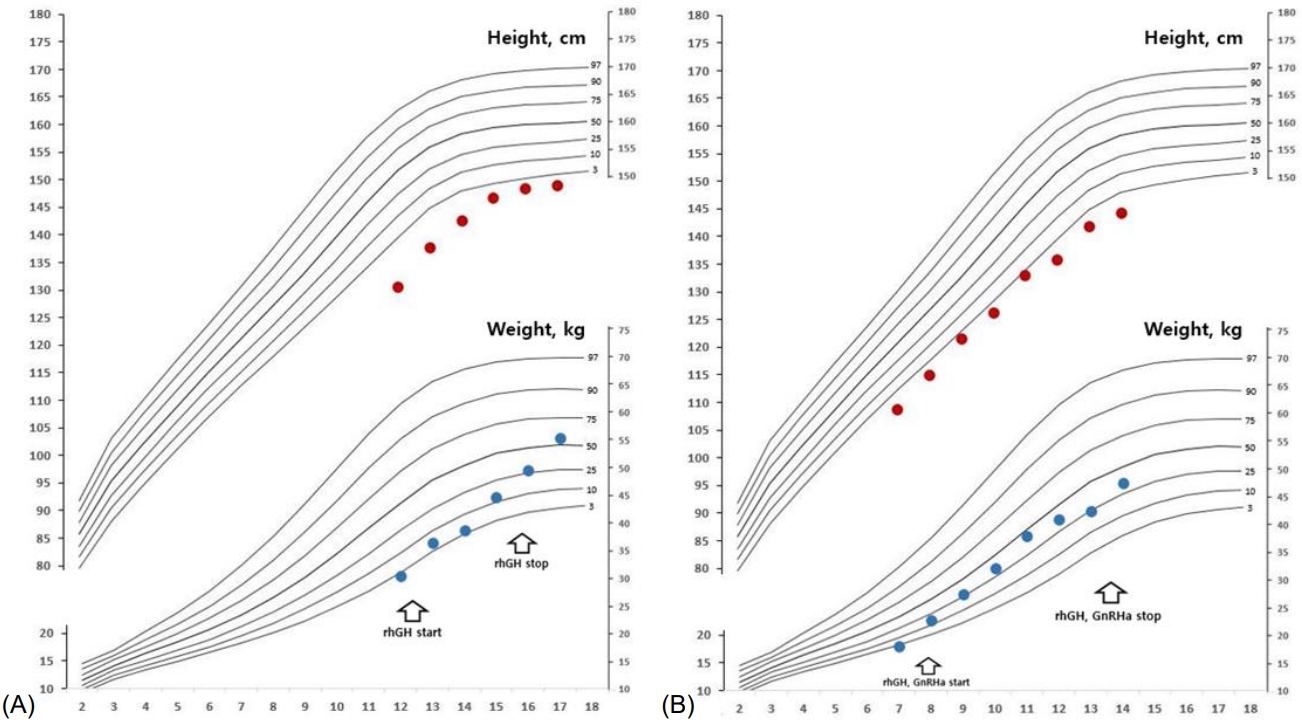
The older sister, an 11-year-old girl whose height was 130.6 cm (-2.28 standard deviation score [SDS]) (case 1), was born at 37+1 weeks of gestation weighing 2,200 g (-1.58 SDS) and was small for gestational age (SGA) after an uneventful pregnancy. No perinatal problems, except microcephaly and ventriculomegaly, were recorded. On physical examination, her head circumference was 47 cm (<-3 SDS), and she had unique craniofacial features including a bird-headed appearance with a receding forehead, micrognathia, and a large beaked nose. Her Tanner stage was 3, and her bone age was the same as her chronological age. Her predicted adult height (PAH) as measured using the Bayley-Pinneau (BP) method was 148 cm. Her karyotype was 46, XX (normal female). Endocrine tests were performed, and her hormone levels were within normal ranges (Table 1). There were no abnormal brain magnetic resonance imaging (MRI) findings. She was started on recombinant human growth hormone (rhGH) treatment at 11 years of age due to no postnatal catch-up growth. Two years after the start of treatment, her scoliosis was aggravated, and she was prescribed a brace. She continued rhGH treatment until she was 15 years old (approximately 4 years into rhGH therapy). At the age of 18 years, her adult height had reached 148.9 cm (-2.47 SDS) (Fig. 1A). She displayed mild cognitive and behavioral disorders during the treatment, for which we consulted colleagues from the department of psychiatry. Her cognitive and intellectual function tests revealed a full-scale intelligence quotient of 80ŌĆō89 according to the Korean Wechsler Adult Intelligence Scale-IV for her age. In the comprehensive attention test, a decrease in interference in attention and divided attention was observed.
2. Case 2
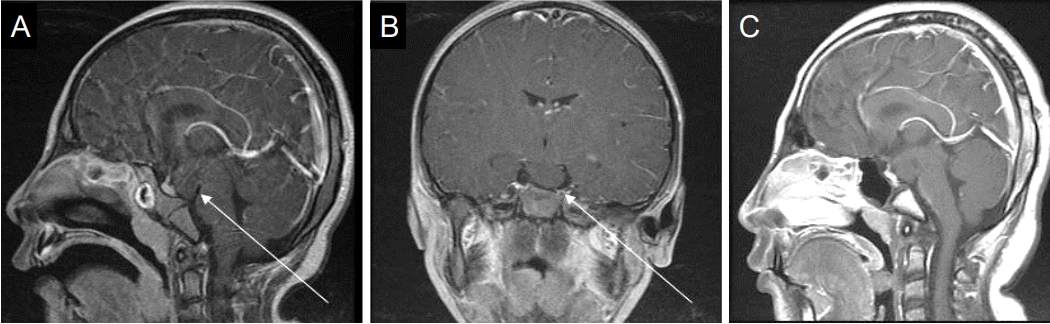
The younger sister, a 6-year-old girl whose height was 108.7 cm (-1.66 SDS) (case 2), was born at 37 weeks of gestation, weighing 2,100 g (-1.61 SDS); she was SGA without other medical problems except microcephaly. On physical examination, she had a small head circumference of 43 cm (<-3 SDS), and both breasts were engorged to Tanner stage IIŌĆōIII. Her bone age was advanced by more than 4 years. Her PAH as measured using the BP method was less than 140 cm. She presented with clinical characteristics similar to those of case 1, including a bird-shaped head with microcephaly, micrognathia, and a large beaked nose. Her karyotype was also 46, XX (normal female). Following endocrine tests, she was diagnosed with CPP and subclinical hypothyroidism (Tables 1, 2). Additionally, brain examination by MRI revealed a HH of a pedunculated nonenhancing mass in the tuber cinereum that extended into the suprasellar cistern (Fig. 2A, B). She received a depot gonadotropin-releasing hormone agonist (GnRHa) with rhGH treatment at 6 years of age for 4 years and 6 months for CPP and growth impairment and also began 50 ╬╝g of concurrent levothyroxine. When she was 11 years old, she slipped on the stairs and was diagnosed with a right-slipped capital femoral epiphysis (SCFE). She underwent surgery for fixation of the right femur head and prophylactic screw fixation of the left capital femoral epiphysis concurrently (Fig. 3A, B). After the surgery, she stopped rhGH and GnRHa and only continued treatment with levothyroxine for hypothyroidism. At the age of 13 years, she experienced menarche. Her height was 144.3 cm (-2.0 SDS) with a bone age of 15 years (Fig. 1B). We followed her HH size using MRI and found no significant interval change (Fig. 2C). She also showed mild cognitive and behavioral disorders and planned to undergo a test to evaluate her attention and behavioral problems.
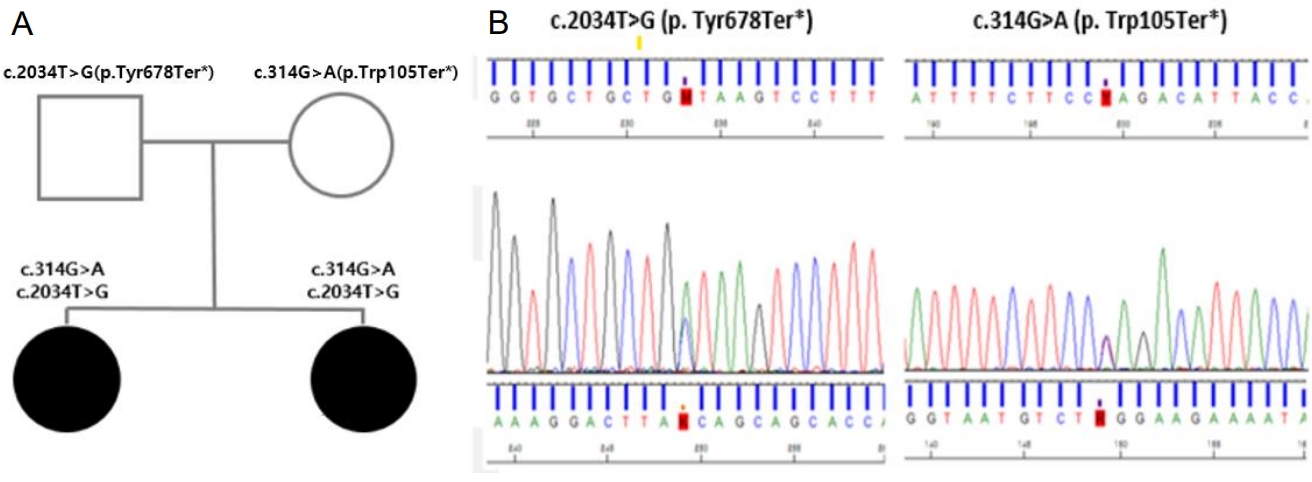
Their parents were healthy and nonconsanguineous. Their motherŌĆÖs height was 168 cm (+1.41 SDS), and their fatherŌĆÖs height was 174 cm (+0.16 SDS). There was no family history of dwarfism, dysmorphic facial features, or developmental delay. Due to the presence of characteristic facial and cranial features, SGA without catch-up growth, HH, SCFE, and behavioral psychiatric problems, we suspected an underlying genetic cause. To identify this, whole exome sequencing was performed and identified 2 compound heterozygous pathogenic variants of the CEP152 gene (c.2034T>G, p. Tyr678Ter* and c.314G>A, p.Trp105Ter*). These variants were inherited from their parents, which was confirmed by Sanger sequencing. SCKL 5 follows an autosomal recessive inheritance pattern, in which genetic disorders occur when an individual inherits 2 nonworking genes for the same trait (one from each parent), and the variant may be either compound heterozygous or homozygous. If an individual receives one working gene and one nonworking gene, the person will be a carrier for the disease but will usually not show symptoms. Thus, their parents were carriers of each variant with no symptoms of SCKL 5. The variant c.2034T>G in CEP152 creates a stop codon at position 678 on the exon, and the variant c. 314G>A creates a stop codon at position 105 on the exon. These variants have been reported to occur at extremely low frequencies in large population cohorts (GenomAD). Both variants were classified as pathogenic according to the recommendations of the American College of Medical Genetics and Genomics [7] and were suspected to cause SCKL 5 with an autosomal recessive inheritance pattern (Fig. 4A, B).
Discussion
Genetic factors may contribute to the etiology of HH [8]. A variant in the GLI3 gene is the genetic cause of Pallister-Hall syndrome in most syndromic cases. Sonic hedgehog gene variants, including GLI3 and PRKACA, account for about onethird of nonsyndromic, sporadic HHs. Recently, a somatic gene related to ciliopathy has been identified as a cause of HH [9]. The association between ciliopathy and centrosome defects has been studied in several human diseases [10].
The reported brain structural abnormalities related to SCKL include gyral hypoplasia, macrogyria, corpus callosum agenesis, cysts, hypoplastic cerebrum or cerebellum, pachygyria, brain herniations, and central vasculopathy [3,6]. The pathogenesis of these abnormalities in SCKL is not fully understood. However, the centrosome plays a crucial role in cell division, and its activity appears to be critical to cerebral and neuronal development. We suggest that HH is a newly discovered manifestation of cerebral cortex abnormalities in SCKL.
According to clinicopathological classifications, HH lesions can be either parahypothalamic (pedunculated) or intrahypothalamic (sessile). While intrahypothalamic HHs are associated with gelastic seizures and neurological issues, parahypothalamic HHs are commonly associated with CPP [2,11]. HH lesions associated with CPP typically have a base of attachment to the inferior surface of the hypothalamus in the tuber cinereum. Case 2 in our study had an MRI finding of parahypothalamic HH and showed only CPP symptoms without neurological problems. Because she had CPP accompanied by severe growth failure, she was treated with a GnRHa and rhGH. At the beginning of the treatment, we were concerned about a potential increase in size of the HH with administration of rhGH and development of subsequent neurological issues. However, this problem did not occur.
SCKL is a rare genetic disorder; fewer than 200 families have been reported [4]. It is an autosomal recessive neurodevelopmental spectrum disorder characterized by intrauterine growth retardation that results in low birth weight and a small head circumference; proportionate osteodysplastic primordial dwarfism; growth retardation; variable intellectual disability; and unique facial features including a beak-like nose, sloping forehead, narrow face, large eyes, and micrognathia [3,4]. To date, there has been only one report on the effect of rhGH treatment on final height in children with SCKL. In that study, the final height of 2 patients who underwent long-term rhGH treatment increased by 7.2 and 3.4 cm, respectively, compared with their siblings who did not receive rhGH treatment and therefore experienced no additional growth [12]. It is difficult to evaluate the effects of rhGH treatment on growth because of the lack of disease-specific growth charts for this very rare genetic syndrome that presents with high heterogeneity. The PAH of case 2 measured using the BP method before treatment was less than 140 cm. However, her current height is greater than 144.3 cm. Therefore, we conclude that she responded well to the rhGH treatment.
SCFE is a hip disorder that presents with separation of the femoral head from the femoral metaphysis and commonly occurs in late childhood and early adolescence [13]. The causes of SCFE have not been identified, although hormonal disorders (including hypothyroidism, hyperparathyroidism, hypogonadism, gigantism, and obesity) and rhGH treatment are known to be related [14,15]. Several studies of the relationship between SCFE and GnRHa treatment have been published [13,15)]. We treated the younger sister described here with concurrent GnRHa and rhGH therapy. She also had SCKL, which is characterized by osteodysplastic dwarfism. Interestingly, although the 2 sisters received the same dose range of rhGH treatment and had the same variant of the CEP152 gene, only the younger sister presented with SCFE. Of note, the accident in which the younger sister slipped down the stairs could have contributed to the occurrence of SCFE in already weakened bones. With the lack of previous case reports of SCFE in patients with SCKL, we could not conclusively determine the reason for SCFE in this patient. However, we propose that a combination of factors, including pubertal age, underlying osteodysplastic syndrome, endocrinopathy (such as CPP and subclinical hypothyroidism), GnRHa and rhGH treatment, and the fall all contributed to the SCFE. We suggest that careful monitoring is required for SCFE in adolescent patients with genetic syndromes that include skeletal involvement and endocrinopathy.
This is the first report of HH related to CPP as a manifestation of SCKL. We were intrigued by the difference in HH occurrence between 2 sisters carrying the same variant of the CEP152 gene. This finding may also imply that the penetrance of variants varies between individuals with SCKL. Additionally, they were the first SCKL patients diagnosed by genetic confirmation in Korea.