Highlights
┬Ę We report the first case of dopamine-secreting pheochromocytoma in an adolescent patient with NF1 in Korea. When metanephrine is negative in patients with a high risk of pheochromocytoma, the possibility of dopamine secreting pheochromocytoma should be considered.
Introduction
Pheochromocytoma (PCC) and paraganglioma (PGL) are rare neuroendocrine tumors in children. PCC is a catecholamine-secreting tumor arising from chromaffin cells in the adrenal medulla [1]. Hereditary PCC occurs with a frequency of 15% in multiple endocrine neoplasia type 2, von Hippel-Lindau syndrome, neurofibromatosis type 1 (NF1), and familial PGL [2]. In children, PCC and PGL as the causes of hypertension occur at a very low frequency of 0.5-2% [3].
NF1 is a genetic disorder characterized by the presence of multiple soft tissue tumors of neural crest origin, caf├®-au-lait spots, Lisch nodules, and dysplastic bone lesions with an incidence rate of 1 in 2,500ŌĆō3,000 individuals [4]. NF1 has a high risk of tumor occurrence [5]. The incidence rate of PCC in adult patients with NF1 is reportedly 0.1%ŌĆō5.7%, of which there is a 20%ŌĆō50% chance of hypertension [6,7]. Moreover, the incidence of PCC in pediatric patients with NF1 is 4%, and most of these tumors secrete epinephrine and norepinephrine [8]. PCCs that do not produce norepinephrine and epinephrine and instead secrete their precursor, dopamine, are extremely rare. Several cases of dopamine-secreting PCCs have been reported worldwide in adults [9-16]; however, such cases have not been reported in children or adolescents.
Herein, we report the first case of a dopamine-secreting PCC in an adolescent with NF1 in Korea based on the results of clinical, biochemical, imaging, and surgical treatments. Dopamine-secreting PCC does not show the typical symptoms of catecholamine excess; as such, the diagnosis can be delayed. In high-risk PCC patients (such as children with NF1), regular checkups, focused examinations, and a multidisciplinary approach are necessary for proper diagnosis and treatment.
Case report
A 13-year-old boy was referred to the endocrinology clinic for further examination of a 2-cm-sized left adrenal gland mass discovered on abdominal computed tomography (CT) during hypertension workup. He was already diagnosed with NF1 and was regularly followed up by the neurology department. Hypertension was found incidentally, and he was hospitalized for further examination. He was not taking any medications.
He was born at 40 weeks via cesarean section and weighed 3,600 g. At 6 years, multiple caf├®-au-lait spots were observed, and he was diagnosed with NF1 based on axillary and inguinal freckles. His father, grandfather, and older brother were diagnosed with NF1, but none had neuroendocrine tumors. There were no specific findings on physical examination other than the multiple caf├®-au-lait spots, freckles, and complaints of occasional headaches.
At admission, the patient presented with intermittent headaches and persistent hypertension but did not complain of typical symptoms of catecholamine excess (i.e., paroxysm of headache, palpitations, and/or diaphoresis). All vital signs were within normal ranges, with the exception of hypertension: blood pressure 150/79 mmHg, heart rate 94 beats/min, body temperature 36.5Ōäā, respiratory rate 20/min, and room air O2 saturation 100%. His height was 173 cm (90%ŌĆō95% percentile), weight was 86 kg (>97% percentile), and body mass index (BMI) was 29.4% percentile (obesity). Adrenal gland-related hormone testing was performed because the findings indicated an adrenal incidentaloma. In the serum catecholamine test for PCC, epinephrine was 30.78 pg/mL (normal range: <50 pg/mL), norepinephrine was 138.8 pg/mL (normal range: 110ŌĆō410 pg/mL), and dopamine was 11.49 pg/mL (normal range: <87 pg/mL). The 24-hour urinary tests revealed epinephrine of 17.58 ╬╝g/day (normal range: Ōēż40 ╬╝g/day) and norepinephrine of 26.32 ╬╝g/day (normal range: Ōēż80 ╬╝g/day). However, the 24-hour urine test for dopamine showed elevated level of 572.24 ╬╝g/day (normal range: 65ŌĆō400 ╬╝g/day) (Table 1). The 24-hour urine tests for metanephrine, vanillylmandelic acid, and homovanillic acid excretion were 0.47 mg/day (normal range: Ōēż0.80 mg/day), 4.85 mg/day (normal range: Ōēż8 mg/day), and 4.9 mg/day (normal range: 1.4ŌĆō8.8 mg/day), respectively.
The basal 24-hour urine-free cortisol test performed to rule out Cushing syndrome revealed a value of 78.3 ╬╝g/day (normal range: 4.3ŌĆō176.0 ╬╝g/day). Cushing syndrome was excluded due to a cortisol level of 2.2 ╬╝g/dL (normal range: Ōēż5 ╬╝g/dL) on overnight dexamethasone suppression test. Liver enzyme, lipid profile, and thyroid function tests showed normal findings. Fasting blood glucose and insulin levels were 95 mg/dL (normal range: 70ŌĆō110 mg/dL) and 27.8 ╬╝U/mL (normal range: 1.6ŌĆō10.8 ╬╝U/mL), respectively. The homeostatic model assessment for insulin resistance was calculated at 6.5, suggesting insulin resistance. The HbA1c level was 5.2%. There were no abnormal findings on 2-dimenstional echography and doppler kidney ultrasound for hypertension.
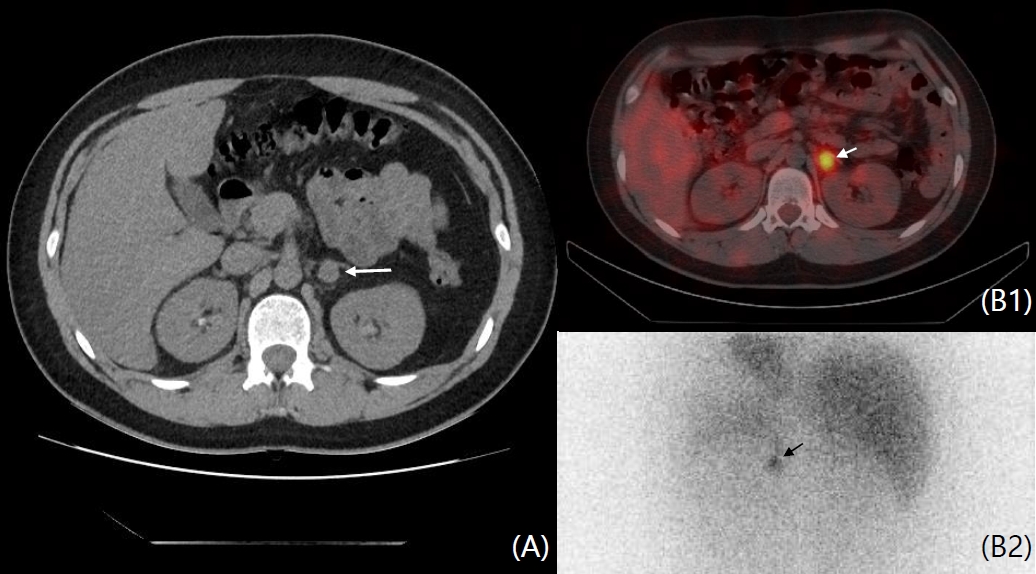
On abdominal CT, a well-defined 1.2├Ś1.4├Ś1.7-cm-sized homogeneous nodular lesion in the left adrenal gland was highly suggestive of lipid-poor adenoma, but PCC could not be ruled out. On 123I-metaiodobenzylguanidine (MIBG) single-photon emission tomography/CT (SPECT/CT), clear MIBG uptake was observed in the nodule on the left adrenal gland for both the 24-hour and 48-hour images, suggesting PCC (Fig. 1).
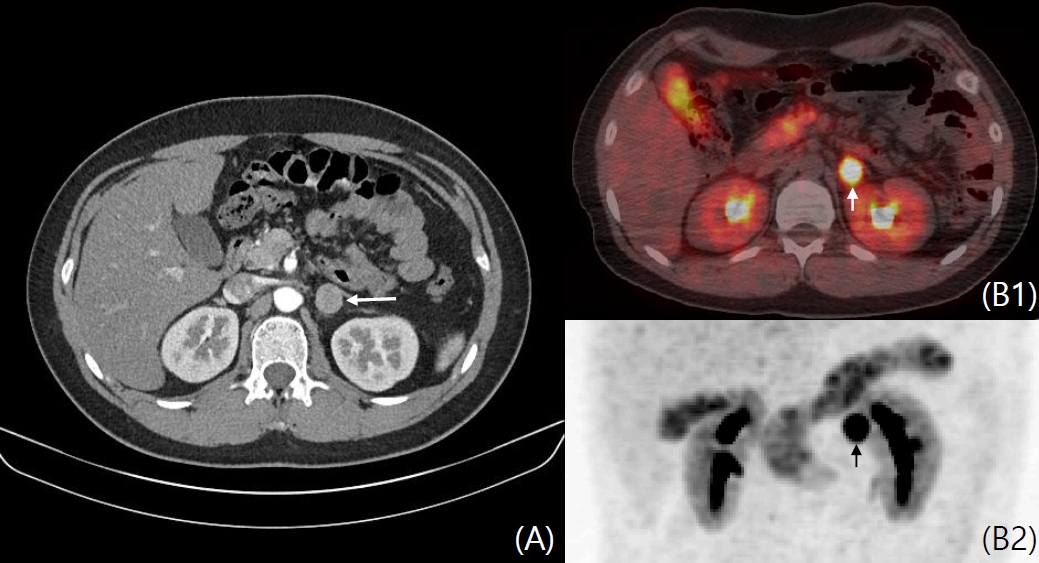
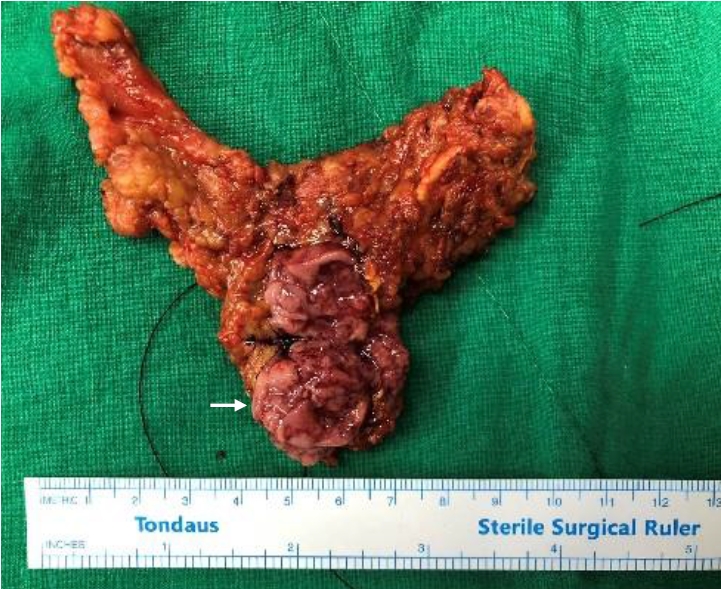
A multidisciplinary conference was held with pediatric endocrinology, radiology, nuclear medicine, and pediatric surgery departments due to the small lesion size, vague symptoms, and biochemical findings with only an increase in urinary dopamine level. Assuming that immediate laparoscopic surgery would be difficult, it was decided to consider surgical treatment after 3-month progress based on regular blood pressure checkups, biochemical tests, and imaging tests. After this period, his BMI improved from 29.4 kg/m2 to 28.0 kg/m2, and his systolic blood pressure (SBP) was 110ŌĆō120 mmHg. After 6 months, there was no significant difference in BMI (28.1 kg/m2), and vital signs showed hypertension, tachycardia with blood pressure of 157/92 mmHg, and a heart rate of 110 beats/min. Fractionated 24-hour urine metanephrine excretion was within the normal range; however, the 24-hour urine dopamine test showed elevated level at 435.53 ╬╝g/day (normal range: 65ŌĆō400 ╬╝g/day) (Table 1). An increase in the size of the adrenal mass was noted on follow-up abdominal CT, and focal significant uptake of 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine (18F-FDOPA) PET/CT was identified in the left adrenal gland (Fig. 2). Laparoscopic resection of the left adrenal mass was performed under suspicion of dopamine-secreting PCC due to increased blood pressure, heart rate, tumor size, and positive FDOPA uptake during follow-up (Fig. 3). Preoperative alpha-blockade was administered, and blood pressure remained stable during surgery without hypotension. During surgery, SBP was 108ŌĆō120 mmHg, diastolic blood pressure was 66ŌĆō81 mmHg, and pulse rate was 63ŌĆō102 beats/min. The postoperative course was uneventful without complications. Immunohistopathologic findings revealed positive synaptophysin, chromogranin, and S-100, indicating PCC. The patient was already diagnosed with NF1, but no mutations were found in either the SDHB or SDHD gene, which are highly associated with dopamine-secreting tumors. One year after surgery, his vital signs, 128/77 mmHg blood pressure, and 74 beats/min heart rate indicated stable conditions. The 24-hour test findings for catecholamine, including 296.56 ╬╝g/day urine dopamine (normal range: 60ŌĆō400 ╬╝g/day) and serum catecholamine level, were all within the normal range (Table 1). There was no uptake on the follow-up 18F-FDOPA PET scan. Regular follow-up of the patient is performed every 6 months.
This study was approved by the Institutional Review Board of Kyungpook National University Chilgok Hospital, Daegu, Korea (approval number: 2021-03-002). Informed case consent was obtained from the patientŌĆÖs parents or guardian for publication of this case report.
Discussion
Here, we present a rare case of PCC wherein the catecholamines commonly secreted by this tumor (norepinephrine and epinephrine) were not produced, and instead dopamine was secreted. Dopamine is a precursor of norepinephrine and is produced early in the biosynthesis of catecholamines [1]. Decreased expression of dopamine beta-hydroxylase, the enzyme that converts dopamine to norepinephrine, has been detected in dopamine-secreting PCC. Without dopamine betahydroxylase, there is an overproduction of dopamine instead of epinephrine or norepinephrine [9]. Dopamine-secreting PCCs show different characteristics from norepinephrine- and epinephrine-secreting tumors, and dopamine causes different reactions depending on plasma concentrations. At low dopamine concentrations, vasodilatation causes hypotension and increased peripheral tissue perfusion. At high dopamine concentrations, beta-1 adrenergic receptors are stimulated to increase cardiac output. At even higher concentrations, alpha-1 adrenergic receptors are stimulated, causing peripheral blood vessel contraction (vasoconstriction), which increases blood pressure [17]. Dopamine-secreting pheochromocytoma-paragangliomas (PPGLs) do not show the typical symptoms of catecholamine excess, and instead patients are usually normotensive. Moreover, non-specific symptoms, such as diarrhea and weight loss, are noted more often, causing delays in diagnosis that allow time for the tumor to increase in size and be detected only due to symptoms of mass effect [10,18].
Foo et al. [10] reported that 20% of patients had symptoms of adrenergic excess, and hypertension was noted in 13.3%; however, none of the patients presented with hypertensive crisis. Proye et al. [11] also reported that hypertension is not present in dopamine-only secreting tumors but is present in combined dopamine-, noradrenaline-, and adrenaline-secreting tumors. Our patient did not show the typical clinical symptoms of PCC, with only hypertension and occasional ambiguous headaches. Moreover, our patient was obese with insulin resistance, and metabolic syndrome as the cause of hypertension could not be excluded. As a result of lifestyle modification, BMI decreased and blood pressure seemed to improve; however, tachycardia reoccurred despite the improved BMI and hypertension. There was an increase in the adrenal mass size, as noted on follow-up abdominal CT, and the 24-hour urine dopamine level was high. Eventually, surgical treatment was performed. Postoperatively, his vital signs were stable without hypertension or tachycardia, the 24-hour urine dopamine was normal level, and PCC was histologically diagnosed.
Plasma-free metanephrine or urinary fractionated metanephrine examination is recommended as the initial biochemical test to diagnose PCC, as it is highly sensitive and specific [18]. As shown in our case and as metanephrine-free PPGL is rare, the tests for dopamine-secreting PCC may not detect an increase in metanephrine level. In such cases, epinephrine, norepinephrine, and 3-methoxy-4-hydroxymandelic acid may be negative, and plasma and/or 24-hour urine dopamine tests may be the only reliable way to detect the tumor [12,13]. The measurement of 3-methoxy-tyramine, a metabolite of dopamine, may also be helpful in diagnosis [14]. Unfortunately, 3-methoxy-tyramine testing was not available at Kyungpook National University Children's Hospital. In our patient, all the biochemical test results, except the 24-hour urine dopamine test, showed negative findings several times.
As a functional imaging modality in patients with metastatic PPGL, 123I-MIBG scintigraphy is recommended. MIBG is an analog of norepinephrine, showing 90% sensitivity and specificity in detecting norepinephrine- and epinephrine-secreting PCCs [18]. Exclusively dopamine-secreting PCC, which does not secrete norepinephrine and epinephrine, may not be detected using 123I-MIBG [12]. Thus, 18F-FDG PET using glucose metabolism or 18F-FDOPA PET using a dopamine precursor can be an alternative [19,20]. In our case, uptake by the suspected lesion increased by initially performing the 123I-MIBG test; however, PCC was not diagnosed because there was no explicit definitive finding on combined CT and PET/ CT images. Hence, for six-month follow-up imaging, PET/CT scanning using 18F-FDOPA, rather than 123I-MIBG, was used. The same region showed increased uptake, supporting the possibility of dopamine-secreting PCC.
The treatment for PCC is surgical removal. However, because hypertensive crisis may occur during surgical treatment for epinephrine- and norepinephrine-secreting tumors, use of an alpha-blocker is mandatory for preoperative management [1,18]. For dopamine-secreting PCC, blood pressure is often within the normal range, and there have been reports of hypotension during alpha-blockade; therefore, preoperative management is not routinely recommended [11]. However, there are previous reports of successful surgical resection after using an alpha-blocker as preoperative management for dopamine-secreting PPGL [15]. In addition, intraoperative hypertensive crisis has been reported in some cases of dopamine-secreting PPGLs, and preoperative alpha-blockade should be considered [16]. In our patient with stage 2 hypertension, he received a preoperative alpha-blocker while closely monitoring blood pressure and then a beta blocker. Surgery was performed because the blood pressure was stable for a sufficient time during preoperative management, as well as during and after the surgery. In the case reported by Yasunari et al. [9], immunohistochemistry revealed a decrease in the dopamine beta-hydroxylase enzyme production when diagnosing dopamine-secreting PCC. Moreover, the only adult case reported in Korea showed increased expression of dopamine in immunohistochemical testing [12]. Unfortunately, immunochemistr y for dopamine beta-hydroxylase and dopamine could not be performed in our study due to lack of availability at our hospital. Metyrosine, a tyrosine analog that blocks dopamine synthesis by inhibiting tyrosine hydroxylase, can be an option for preoperative control of symptoms of these tumors; however, this test was also not available at our hospital [13].
In conclusion, this study reports a case of dopamine-secreting PCC in an adolescent patient with NF1 in Korea. For patients with a high risk of PCC, such as children with NF1, it is recommended to consider a diagnosis of dopamine-secreting PCC when metanephrine results are negative, although this is not applicable for all adrenal incidentalomas. Additionally, if symptoms and imaging are ambiguous, regular follow-ups, 18F-FDOPA PET-focused examinations, and a multidisciplinary approach are required.