Highlights
┬Ę MODY type 4 is an infrequent form of monogenic diabetes brought on by heterozygous mutations in the PDX1 gene. We report the first case of MODY type 4 phenotype in Korea and should consider using genetic analysis for precise diagnosis with MODY.
Introduction
Diabetes mellitus (DM) in children and adolescents has increased worldwide and is a heterogeneous disease, with most cases corresponding to type 1 and type 2 DM [1]. Although type 1 DM is the most common form in pediatric patients, type 2 DM and monogenic diabetes have also been detected [2].
Monogenic diabetes can include maturity-onset diabetes in youth (MODY), neonatal diabetes, and mitochondrial diabetes, resulting from mutations in genes encoding transcription factors or other proteins that regulate pancreas development or function [3]. MODY is one of the most common types of monogenic diabetes, accounting for less than 5.0% of all patients with DM [4] or 1%ŌĆō6% of patients with DM [2]. Generally, MODY is characterized by an autosomal dominant inheritance that can be transmitted by either parent or occurs as a de novo mutation. It is classically characterized by a nonacute and nonketotic presentation, typically occurring before 25 years of age [5]. As patients with MODY rapidly fail treatment with oral drugs or do not present with ketosis, they can be classified as type 1 DM or type 2 DM [5].
MODY type 4 is generally caused by heterozygous mutations in the pancreas and duodenum homeobox-1 (PDX1) gene, a pancreatic homeodomain transcription factor that is critically involved in both early pancreatic development and the regulation of ╬▓-cell function [6]. MODY type 4 is a rare form that presents a few cases of PDX1 mutations [6]. Clinical presentation of MODY type 4 patients includes a variable age of onset with obese or nonobese phenotype and is managed with diet, oral antidiabetic medications, and/or insulin [4].
In Korea, MODY cases have been reported in 11 studies, with examples of the most common forms of glucokinase (GCK)-MODY, hepatocyte nuclear factor-1 homeobox A (HNF1A)-MODY, and hepatocyte nuclear factor-4 homeobox A (HNF4A)-MODY [7]. PDXI mutation-associated MODY type 4 is extremely rare in Korea; here, we describe the first case of a Korean girl who was initially diagnosed with type 2 DM.
Case report
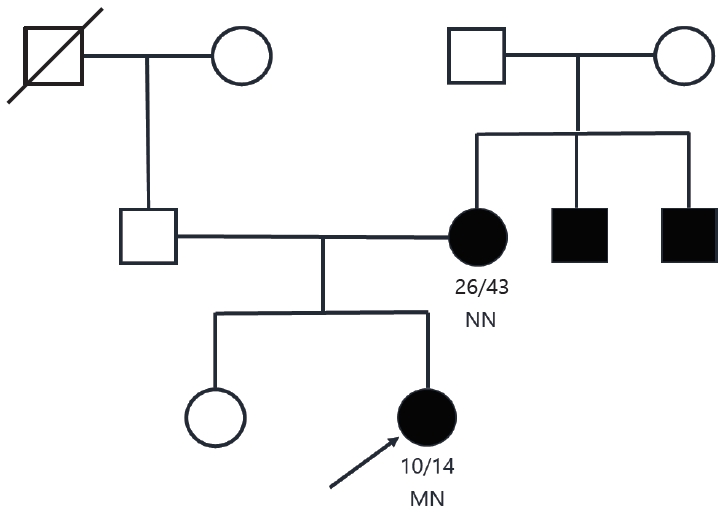
A 10-year-old girl visited our outpatient clinic for hyperglycemia as a result of a serum glucose test performed at a local pediatric clinic due to glucosuria detected during a school checkup. At the first visit, she was nonobese, her height was 146.3 cm (50thŌĆō75th percentile for age), bodyweight 32.4 kg (25thŌĆō50th percentile for age), and body mass index 15.14 kg/m2 (10thŌĆō25th percentile for age). Her mother had been diagnosed with type 2 DM at the age of 34 years and had developed gestational DM at the age of 26 years with no diabetic complications. At diagnosis of type 2 DM, her motherŌĆÖs body mass index (BMI) was 20.3 kg/m2 and glycated hemoglobin (HbA1c) level was 12.9%. Her mother was being effectively controlled with oral antidiabetic medication. Additionally, her 2 maternal uncles had been recently diagnosed with type 2 DM. Her father and elder sister both were nondiabetic. The patientŌĆÖs pedigree is shown in Fig. 1.
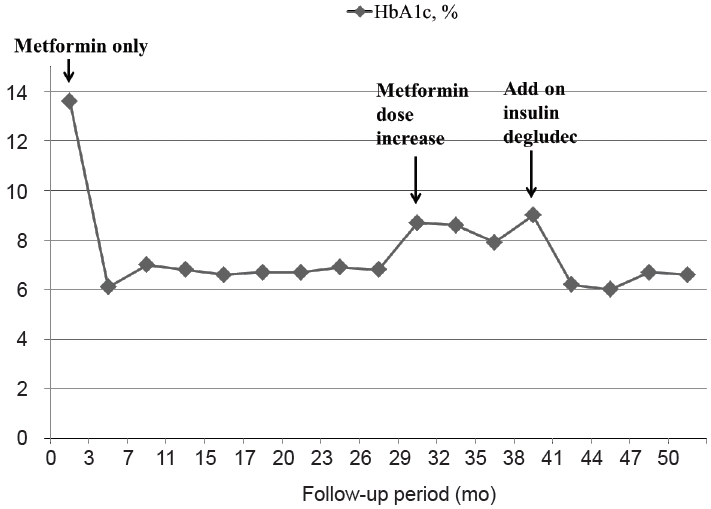
The initial fasting plasma glucose (FPG), HbA1c, fasting C-peptide, and 2-hour postprandial C-peptide after meal levels were 159 mg/dL, 13.6%, 0.84 ng/mL (normal range, 0.8ŌĆō4.2 ng/mL), and 24.44 ng/mL, respectively. No islet-related autoantibodies, including insulin autoantibodies, antiglutamic acid decarboxylase antibodies, or anti-islet cell antibodies, were detected in the blood. Based on these results, the patient was diagnosed with type 2 DM and received metformin monotherapy. Her serum glucose level was well managed, and her HbA1c level decreased to <7.0% after treatment. However, after 2 years of metformin monotherapy, her serum glucose level deteriorated, and her HbA1c level increased to 9.0%, although there were no changes in body weight and lifestyle, including diet. She was admitted to our hospital for hyperglycemic management and workup.
Upon admission, her height was 154 cm (10thŌĆō25th percentile for age), bodyweight 41.6 kg (10thŌĆō25th percentile for age), and body mass index 17.54 kg/m2 (10thŌĆō25th percentile for age). Other physical examinations were common. FPG level was 163 mg/dL and HbA1c was 9.0%, with a fasting C-peptide level of 1.08 ng/mL and a 2-hour postprandial C-peptide level of 1.47 ng/mL. No other abnormal liver or kidney function was detected.
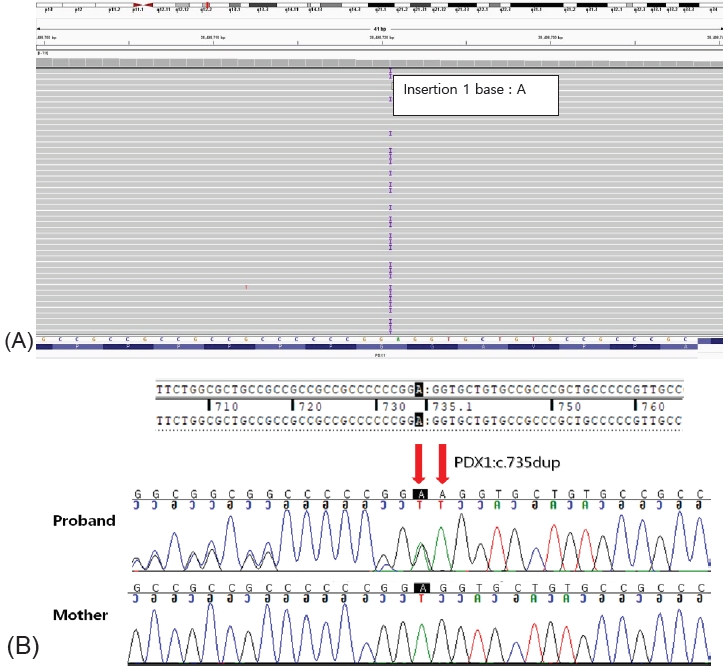
Based on these findings, a slight decrease in endogenous insulin secretion combined with early onset, nonobesity, antibody-negativity, and a family history of diabetes, MODY was suspected. Genetic tests were performed after obtaining written informed consent of the patient and her mother. Screening of MODY-related genes using next-generation sequencing (NGS) panel analysis detected the PDX1 variant, a heterozygous duplication of c.735dup (p.Gly246ArgfsTer21) in exon 2 of the PDX1 gene (Fig. 2), which was diagnosed as MODY type 4 and was not reported. Based on the 2015 guidelines developed by the American College of Medical Genetics and Genomics/Association for Molecular Pathology for classification of pathogenic or likely pathogenic variants, the PDX1 mutation identified in our proband was "likely pathogenic" [8]. The variant results in a glycine (Gly) to arginine (Arg) substitution at position 246, which results in a premature termination codon. Her mother's sample was also subjected to NGS, and no mutations were detected. The clinical and laboratory data of this Korean family are presented in Table 1.
Insulin therapy was added to metformin monotherapy because of the potential decrease in insulin secretion as the pancreatic function might be gradually reduced. The patient commenced treatment with metformin and insulin degludec combination therapy.
After hospital discharge, the patient was treated with metformin and insulin combination therapy. Based on follow-up examinations, HbA1c was maintained below 7%, and no comorbidities were found; therefore, this combination therapy was continued and adjusted according to changes in the serum glucose and HbA1c levels (Fig. 3).
This study was approved by the Institutional Review Board of Gil Medical Center (approval number: GCIRB2021-324). The authors have informed the patient of the use of data and clinical pictures for publication purposes.
Discussion
MODY is an autosomal dominant inherited disorder that presents early in life due to defects in the development of optimum ╬▓-cells or failure in their function. At least 14 types of MODY that are diagnosed in diabetes patients are caused by mutations in several essential genes, such as GCK, HNF1A, hepatocyte nuclear factor-1 homeobox B (HNF1B), and PDX1 [9].
Recently, MODY was suspected in a patient with a family history of diabetes in 1 parent and first-degree relatives of the affected parent, with lack of characteristics of type 1 DM, including the absence of autoantibodies, a small requirement for insulin supplementation, and lack of type 2 DM characteristics, which is distinct from obesity and acanthosis nigricans [2]. In addition, in the case of patients with diabetes in the first 6 months of life, molecular neonatal DM (NDM) screening should immediately be conducted [2]. MODY is often misdiagnosed as type 1 or type 2 DM, and it is estimated that > 80% of MODY cases are not diagnosed by molecular testing, as in the present case [10].
In the present case, progressive hyperglycemia was observed for several years due to a mutation in PDX1. Initially, the disease was misdiagnosed as type 2 DM because of her clinical features, where the lack of pancreatic islet autoantibodies was not detected, stimulated serum C-peptide level was higher than 0.6 ng/mL, and there were no other endocrine diseases. Although she had been treated with metformin since she was diagnosed with type 2 DM, the patient abruptly presented with hyperglycemia without any evidence of a trigger. There is a need for reinvestigation that aids the diagnosis of either type 1 DM or MODY. Genetic analysis revealed a mutation in PDX1, which is associated with MODY type 4.
MODY type 4 is a rare form of monogenic diabetes caused by heterozygous mutations in the PDX1 gene, which encodes the transcription factor PDX1 crucial for pancreatic and beta-cell development and function. Homozygous mutations in PDX1 result in pancreas agenesis associated with NDM [6,11]. However, heterozygous mutations in PDX1 are associated with DM in humans, suggesting that PDX1 plays a role in islet compensation for insulin resistance or glucose-stimulated insulin release, particularly in postprandial hyperglycemia [6]. Caetano et al. reported a heterozygous PDX1 variant that revealed caudal pancreatic agenesis and no main pancreatic duct of this segment on computed tomography [12].
Duplication in exon 2 of the PDX1 gene c.735dup has not been detected in Korea. The proband's mother, who had been diagnosed with type 2 DM, did not have a PDX1 gene mutation. Her father, who was nondiabetic, did not undergo a genetic analysis; thus, we assumed that our proband was affected by a de novo mutation, which is considered rare [13]. Nevertheless, recent epidemiological studies have demonstrated that such mutations in the major MODY genes, such as GCK, HNF1A, and HNF4A, could be more frequent than previously assumed [13].
Various clinical characteristics of patients diagnosed with MODY type 4 have been described in several studies (Table 2). The age at onset is distributed widely from neonatal to 26 years, with a mean age of 10 years. Most patients test negative for ╬▓-cell antibodies. HbA1c of previous patients with available data at diagnosis of MODY ranged from 7.4%ŌĆō10.7%, all diabetic HbA1c level Ōēź6.5%. However, our proband's BMI was lower than that reported in other cases. Unfortunately, it is not possible to compare each clinical feature due to insufficient patient information. Further detailed case reports demonstrating disease progression and complications are necessary to improve the existing knowledge of MODY type 4.
Therapeutic management of MODY type 4 has not yet been established because the traditional criteria of MODY identify a low proportion of patients and are not sufficiently sensitive to be used alone in clinical practice [14]. Nevertheless, the effectiveness of oral antidiabetic medications, such as metformin and dipeptidyl peptidase 4 inhibitors, has been reported in MODY type 4 cases [14-16]. Some cases reported the use of insulin as a primary therapeutic agent [17]. Al-Kandari et al. [18] found a missense variant of c.461C > G (p. Thr154Arg) in a 16-year-old Kuwaiti proband who was receiving insulin treatment. In that case, the switch from treatment with metformin only to insulin combination treatment with metformin was successful and led to reasonable control of glycemia.
A correct genetic diagnosis impacts the treatment and identifies at-risk family members. Thus, it is essential to consider a diagnosis of MODY in appropriate individuals and to pursue genetic testing to establish a molecular diagnosis. Molecular analysis using targeted-NGS provides simultaneous analysis of the genes in a single panel at a cost comparable to Sanger sequencing [19].
The present case had some limitations. We were not able to test the patientŌĆÖs father, sibling, or 2 maternal uncles for the detected gene mutation to determine whether the mutation arose de novo.
To the best of our knowledge, there have been no reports of PDX1-MODY in Korea [7]. This report describes the first such case of MODY type 4, caused by a heterozygous PDX1 mutation c.735dup (p.Gly246ArgfsTer21). This emphasizes the diversity of clinical manifestations of MODY type 4 and the importance of genetic screening and counseling in these patients. Genetic and personalized diagnosis and treatment are crucial for young patients with nonclassical forms of diabetes.