Bone health in pediatric patients with neurological disorders
Article information

Abstract
Patients with neurological disorders are at high risk of developing osteoporosis, as they possess multiple risk factors leading to low bone mineral density. Such factors include inactivity, decreased exposure to sunlight, poor nutrition, and the use of medication or treatment that can cause lower bone mineral density such as antiepileptic drugs, ketogenic diet, and glucocorticoids. In this article, mechanisms involved in altered bone health in children with neurological disorders and management for patients with epilepsy, cerebral palsy, and Duchenne muscular dystrophy regarding bone health are reviewed.
Introduction
Osteoporosis is an important comorbidity in children with genetic disorders predisposing to bone fragility and children with acute and chronic illnesses [1]. Osteoporosis in children results from inadequate bone accrual due to diminished rates of bone mineral deposition, whereas osteoporosis in adults primarily occurs from bone mineral loss [2]. Bone mineralization occurs throughout childhood and peaks at the end of adolescence, and the bone mineral density (BMD) achieved during childhood and adolescence determines the risk for osteoporosis and fractures during adulthood [3]. Children lacking adequate bone accretion are at higher risk of developing both fragility fractures during childhood and involutional osteoporosis as adults [4].
Children with neurological disorders such as epilepsy, cerebral palsy (CP), and Duchenne muscular dystrophy (DMD) are at higher risk of low BMD and associated osteoporosis and fractures [5-7]. Pediatric neurological disorders constitute a major secondary cause of osteoporosis in children, as shown in Table 1, and account for a significant portion of the children who are referred for bone density assessment [1,8,9]. In this article, we review the risk factors of low BMD in children with neurological disorders in relation to bone physiology, with a focus on epilepsy, CP, and DMD, and discuss recent developments in management of bone health in these children.

Physiology
1. Bone formation and absorption
The rates of bone absorption and deposition are equal in nongrowing bones, and this balance is maintained through the activities of osteoblasts and osteoclasts, respectively [2]. There is continual bone formation and absorption in living bones, with 4% and 1% of living bone surfaces harboring active osteoblasts and osteoclasts, respectively, at any given time [2]. The balance shifts toward increased bone formation in direct proportion to the amount of stress placed on the bone, with bone mass increasing in response to heavy impacts or loads [2]. As a result, healthy load-bearing bones gain adequate strength to carry heavy loads without risk of fragility fractures [10].
2. Bone mineralization
Calcium and vitamin D play critical roles in mineralization of bone. Most vitamin D is produced naturally in the skin from exposure to sunlight, and less than 10% is obtained orally from consumption of vitamin D-rich foods, such as oily fish (e.g., salmon, mackerel) or supplements (e.g., fish liver oils) [11,12]. The vitamin D produced from sunlight or digestively absorbed is biologically inert and requires activation by two sequential hydroxylation reactions, the first occurring in the liver and the second in the kidneys. In the liver, vitamin D-25-hyroxylase, a cytochrome P450 enzyme, converts vitamin D to 25-hydroxyvitamin D (25(OH)D) (i.e., calcidiol), and this compound is subsequently converted in the kidneys by 25-hydroxyvitamin D3 1-alpha-hydroxylase to 1,25-dihydroxyvitamin D (1,25(OH)2D) (i.e., calcitriol); it is this final form that is biologically active (Fig. 1) [13,14].
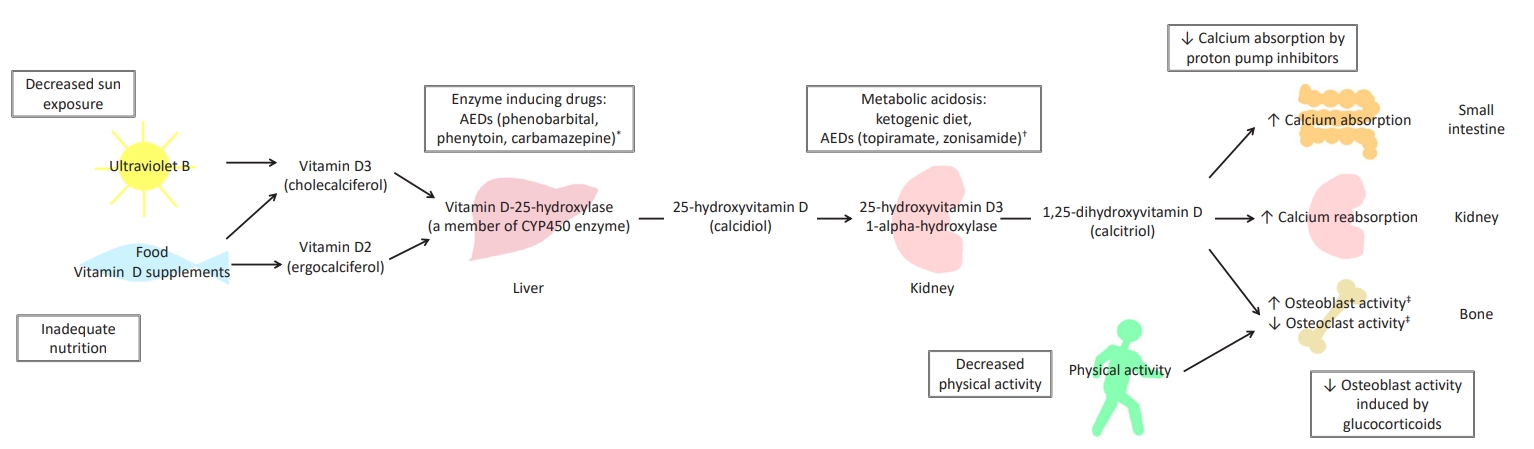
Vitamin D, bone metabolism, and alteration in patients with neurological disorders. Boxed phrases refer to factors that can cause osteoporosis in patients with neurological disorders. * CYP450 enzyme-inducing drugs increase the metabolism of vitamin D resulting in decreased serum 25-hydroxyvitamin D level and subsequently decreased serum 1,25-dihydroxyvitamin D (1,25(OH)2D) level. † Ketogenic diet and other drugs that induce metabolic acidosis cause hypercalciuria in association with calcium loss from bone, resulting in negative calcium balance. Ketogenic diet can also cause inadequate calcium and vitamin D intake. ‡ Such a phenomenon is observed in a setting with normal serum calcium level. In the presence of low serum calcium level, 1,25(OH)2D induces bone resorption. AEDs, antiepileptic drugs.
Vitamin D plays a role in bone mineralization by maintaining adequate serum levels of calcium and phosphorus, which allow osteoblasts to build bone matrix [2]. The active form of vitamin D promotes calcium and phosphorus absorption in the small intestine, calcium reabsorption in the kidneys, increased osteoblast activity, and reduced osteoclast activity [13]. However, 1,25(OH)2D can also enhance bone resorption in the presence of reduced calcium balance [13]. Parathyroid hormone (PTH) regulates production of 1,25(OH)2D in a manner dependent on serum calcium level, as it promotes tubular reabsorption of calcium, increases renal excretion of phosphorus, and stimulates further production of 1,25(OH)2D [15,16].
Mechanisms involved in altered bone health in children with neurological disorders
1. Nutrition
Proper bone growth in an individual necessitates sufficient calcium consumption. Calcium is required for adequate mineralization of bone matrix, and a positive correlation exists between calcium intake and bone mass in individuals of all ages [17,18]. The recommended dietary calcium intake levels in children are 200 mg/day for ages 0–6 months, 260 mg/day for ages 7–12 months, 700 mg/day for ages 1–3 years, 1,000 mg/day for ages 4–8 years, and 1,300 mg/day for ages 9–18 years [19]. Vitamin D is important for calcium absorption, and vitamin D deficiency results in bone demineralization [7]. The major source of vitamin D is obtained by synthesis in the skin from sun exposure, but dietary intake is also important [20]. A serum 1,25(OH)2D concentration of 70–80 nmol/L is generally required to maintain bone health and reduce bone fragility [21]. Vitamin D deficiency also causes muscle weakness, which can be a substantial problem in children with neurological disorders [22,23]. The optimal doses of supplemented vitamin D for children with motor disabilities and generally low serum 1,25(OH)2D concentration are unknown, but vitamin D supplementation at 400 IU/day during infancy and 600–1,000 IU/day during childhood may be needed to prevent secondary osteoporosis [6,19,24,25].
Children with neurological disorders are at greater risk of poor nutritional intake of calcium and vitamin D; furthermore, problems related to nutrition, such as malnutrition, food intolerance, swallowing problems, gastrointestinal reflux, constipation, eating disorders, and drug-nutrient interactions, are more common in these children [26,27].
2. Physical activity
The bone is sensitive to external stimuli and is highly responsive to the mechanical stresses exerted by gravity and muscle contractions [28]. This mechanical loading is necessary to stimulate bone deposition and to maintain skeletal integrity [28]. The forces applied on bones by daily physical activities induce bone remodeling, and this response increases the strength of the bones to endure these stresses [10]. Conversely, inactivity, low muscle support, and conditions of weightlessness have negative effects on bone integrity and cause loss of bone density [28,29], as immobilization and non-weight-bearing situations cause increased osteoclastic activity and cytokine production that results in reduced trabecular formation and thinning of long bone cortices [30].
Children with physical disabilities involving limited ambulation and reduced muscle mass, for example, patients with CP or neuromuscular disease, have lower BMD than healthy children [7,31]. These children can also face a major complication of repeated acute or temporary immobilization following orthopedic surgery or casting to treat fragility fractures; such immobilization causes further reduction in BMD and perpetuates a vicious cycle [32]. Hip casting in children with CP is associated with increased risk of future fracture, and even normal children suffering uncomplicated lower limb fractures casted for more than four weeks have been shown to acquire small, persistent deficits in bone density [33-36]. Separately, adolescents with epilepsy and lacking major motor or sensory impairments were shown to engage in fewer physical activities than sibling controls [37].
In addition to the direct effects of reduced mechanical stress on bone health, the reduced physical activity of children with neurological disorders results in less time spent outdoors, decreased exposure to sunlight, and, consequently, reduced vitamin D synthesis [6].
3. Medications
Several medications commonly used in children with neurological disorders have negative effects on bone health (Table 2). Glucocorticoid treatment is a main therapy in patients with DMD, and glucocorticoids are also used as antiseizure medications. Glucocorticoids impair differentiation of mesenchymal cells into osteoblasts and stimulate an increase in apoptosis of mature osteoblasts, resulting in a decrease in total osteoblast number [38]. In this way, glucocorticoids reduce the function of osteoblasts directly and indirectly by inhibiting expression of insulin-like growth factor I [38]. Glucocorticoids cause an initial increase in osteoclastogenesis but eventually cause a decrease in osteoclastogenesis due to reduced osteoblastic signals [38]. In summary, acute exposure to glucocorticoids enhances bone resorption, and prolonged exposure causes decreased bone remodeling, low BMD, and long-term increased fracture risk [38].
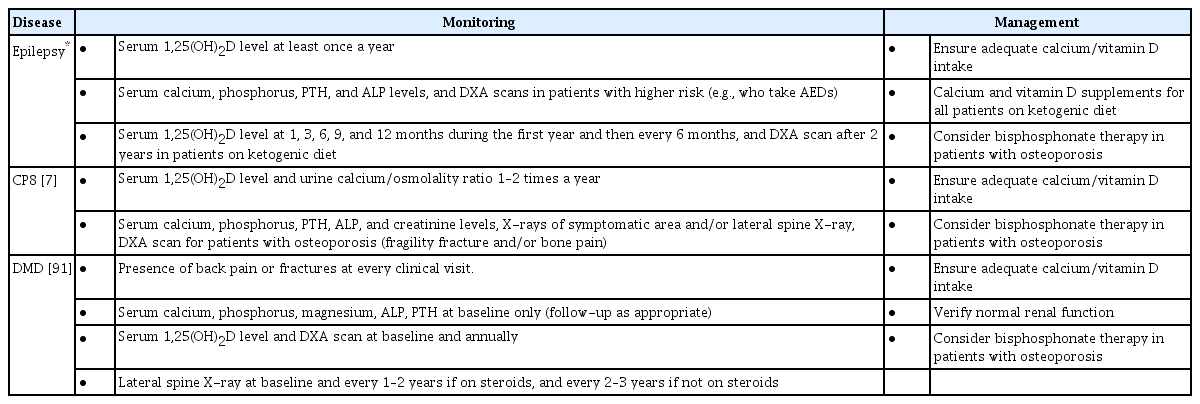
Osteoporosis monitoring and management for patients with neurological disorders
Antiepileptic drugs (AEDs) also have adverse effects on bone health. Inducers of the cytochrome P450 (CYP450) family of enzymes, (i.e., enzyme-inducing AEDs), such as phenobarbital, phenytoin, primidone, and carbamazepine, activate the CYP450 pathway, leading to accelerated vitamin D metabolism with resultant low serum 1,25(OH)2D level, elevated bone turnover, and secondary hyperparathyroidism [39,40]. However, CYP450 enzyme inhibitors such as valproic acid are also associated with low BMD, indicating that activation of the CYP450 pathway and the resultant reduction in vitamin D level is not the only mechanism underlying the AED-associated decrease in BMD [41].
Several studies have shown that benzodiazepines, such as diazepam, lorazepam, midazolam, and clonazepam, are associated with reduced BMD, reduced 1,25(OH)2D level, and increased alkaline phosphatase (ALP) level [42,43]. The overall levels of calcium, phosphorus, magnesium, and PTH were not affected [42,43]. However, these results remain controversial [44,45].
Phenobarbital is an enzyme-inducing AED that causes induction of hepatic microsomal enzymes, resulting in increased 1,25(OH)2D catabolism and osteomalacia [42,46]. Phenobarbital is also associated with direct inhibition of intestinal calcium absorption [47].
Phenytoin is another enzyme-inducing AED and has been reported to be associated with hypocalcemia, hypophosphatemia, the direct inhibition of intestinal calcium absorption [41,48,49].) Phenytoin also inhibited the cellular response and impaired the osteoblastic response to PTH [50,51]. Both primary and secondary hyperparathyroidism were observed in patients receiving phenytoin, and both conditions can result in increased bone resorption [52,53]. However, phenytoin has also been demonstrated to inhibit proliferation of human osteoblast-like cells, suggesting a direct effect on bone metabolism [51,54,55]. Finally, phenytoin exerts an inhibitory effect on calcitonin secretion, resulting in calcitonin deficiency [56,57].
Carbamazepine, another enzyme-inducing AED, causes decreased serum level of 1,25(OH)2D via increased 1,25(OH)2D catabolism, secondary hyperparathyroidism, and increased bone turnover [41]. Carbamazepine also directly inhibits human osteoblast-like cells, causing reduced bone cell proliferation [54], and directly inhibits intestinal calcium transport, although it does so through a vitamin D-independent mechanism [58].
Long-term use of valproic acid has been shown to be associated with low BMD in a dose-responsive manner [41]. The effect of valproic acid on BMD cannot be explained by altered vitamin D metabolism alone, as valproic acid is not an inducer of the CYP450 system but an inhibitor. Instead, valproic acid is believed to lower BMD through stimulation of osteoclast activity, causing an imbalance that contributes to bone loss [59]. However, reports on the mechanisms by which valproic acid causes low BMD show conflicting results. Some studies showed that serum 1,25(OH)2D level was within the normal range, whereas others found low 1,25(OH)2D level [52,60-67]. In most of the studies, serum PTH level was within the normal range; however, in a few studies, a marked increase in PTH level was observed [52,60-62,64-66,68-70]. The results regarding ALP and osteocalcin levels also differed among studies [59,61,62,65,69,71-74]. Valproic acid was not associated with abnormalities in serum calcium or phosphorus level in most of the studies [62,65,66,68,71,75].
The effects of newer AEDs on BMD have not been sufficiently studied. Based on a few studies, lamotrigine and levetiracetam do not seem to be associated with low BMD, whereas an association with low BMD does appear to exist for oxcarbazepine, topiramate, and zonisamide [41]. However, further studies are needed. One mechanism that has been proposed to explain the effects of 2 carbonic anhydrase inhibitors, namely, topiramate and zonisamide, on low BMD is medicationinduced chronic metabolic acidosis that leads to decreased absorption of vitamin D and consequent activation of PTH [6].
4. Ketogenic diet
A ketogenic diet is a dietary treatment used to control seizures in epilepsy patients who are resistant to drug therapies. The classic ketogenic diet uses a 4:1 or 3:1 ratio of fat to nonfat, as measured in grams, such that approximately 80%–90% of the total energy (calorie) obtained from food comes from fat. Patients on a ketogenic diet have been shown to have low BMD, and there was a significant association between low BMD and duration of dietary therapy [76]. Ketogenic diet-induced ketosis causes a chronic ketoacidotic state, which leads to decreased absorption of vitamin D, secondary activation of PTH, and increased bone resorption [6]. The high incidence of renal calculi and the elevated urine calcium:creatinine ratio observed in patients on a ketogenic diet also indicate a major effect of the ketogenic diet on calcium metabolism [76].
Management
1. Epilepsy
More than 50% of patients with epilepsy have low BMD, and the frequency of fractures in pediatric epilepsy patients is 2 to 3 times higher than that in children without epilepsy [77-80]. The low BMD in epilepsy patients is due to inactivity resulting from comorbidities such as CP or from epilepsy itself, decreased exposure to sunlight resulting from inactivity, and use of AEDs that lower BMD.
Despite the accepted association between epilepsy and low BMD, especially with regard to the impact of AEDs on BMD, not all epileptologists routinely screen for bone health [6,81]. A study showed that only 41% of pediatric neurologists and 28% of adult neurologists routinely evaluate bone health in epilepsy patients taking AEDs, suggesting a need for increased awareness [81].
There are currently no guidelines regarding management of bone health in pediatric epilepsy patients taking AEDs. However, some authors recommend screening serum 1,25(OH)2D level in these patients at least once per year, with more frequent (i.e., monthly) screening if abnormal 1,25(OH)2D level is detected [6,16]. In patients at higher risk of abnormal bone health, such as those who take AEDs known to adversely affect BMD, evaluations of other markers, such as serum calcium, phosphorus, PTH, and ALP levels, should be considered [6]. The specific indications for dual energy X-ray absorptiometry (DXA) scans in pediatric epilepsy patients have not yet been established. However, as current recommendations for DXA scans include patients at high risk for low bone density, DXA scans should also be considered for epilepsy patients who are a high-risk for low bone density score [82].
According to the guideline for management of children on ketogenic diets, serum vitamin D level, along with other markers, should be assessed before starting ketogenic dietary therapy and routinely thereafter, specifically, at 1, 3, 6, 9, and 12 months during the first year and every 6 months for the duration of the diet [83]. A DXA scan is also recommended for all patients after 2 years on a ketogenic diet [83]. Administration of calcium and vitamin D supplements is recommended for all patients, and higher doses of vitamin D supplementation are recommended for patients with low vitamin D level [83].
2. Cerebral palsy
A study showed that 77% of children with moderate to severe CP and 97% of non-ambulatory children had osteopenia, which is defined as a femoral BMD z-score less than -2.0 [5]. In a large population study of 763 children with CP, fracture prevalence was 12%, and more than 70% of fractures occurred in lower limb bones [84].
The cause of low BMD in CP patients is likely multi-factorial. The contributing mechanical factors include limitation of weight-bearing ambulation and a higher incidence of temporary immobilization associated with orthopedic surgery, which occurs commonly in pediatric CP patients [5]. Outdoor activities are also significantly limited in CP patients, resulting in decreased exposure to sunlight and consequent low serum vitamin D level [5]. Oral-motor dysfunction can increase feeding difficulties in children with CP, and this can lead to poor nutrition and low calcium intake [85]. Many children with CP have comorbid epilepsy and take AEDs that often have adverse effects on bone health [5]. In addition to AEDs, the proton pump inhibitors (PPIs) that are widely used in children with CP because of dyspepsia or gastroesophageal reflux also have a negative effect on BMD, as they decrease calcium and magnesium absorption [7]. CP is also often associated with premature birth, and rickets of prematurity occurs more frequently in CP patients [5].
A guideline for children with CP at risk of osteoporosis recommends the following for all patients: routine evaluations for serum 1,25(OH)2D level and urine calcium/osmolality ratio, maintenance of adequate calcium intake and vitamin D supplementation, and encouragement of weight-bearing activities through physiotherapies [86,87]. In CP patients with osteoporosis who have suffered fragility fractures or bone pain, additional tests including analysis of serum calcium, phosphorus, PTH, and ALP levels; lateral spine and wrist X-rays; and DXA scans are indicated, and bisphosphonate therapy should be considered [86,87].
3. Duchenne muscular dystrophy
Boys with DMD who are treated with glucocorticoids frequently develop osteoporosis, which manifests as fragility fractures of vertebrae or long bones [88]. Approximately 20%–60% of boys with DMD experience low-trauma extremity fractures, and 30% experience symptomatic vertebral fractures [88-90]. As vertebral fractures are frequently asymptomatic, their prevalence may be higher than indicated in these previous reports [91].
The main risk factors of osteoporosis in patients with DMD are progressive myopathy causing muscle mass loss, muscle weakness, and inactivity or immobilization and use of glucocorticoids [92]. Glucocorticoids are a main therapy in boys with DMD; administration is started when the patient reaches the plateau phase of motor skills, usually at 4–8 years of age, is maintained continuously for the life of the patient [93]. There have been several studies suggesting that deflazacort has fewer adverse effects on BMD than does prednisolone or methylprednisone, but other studies have been unable to confirm the potential bone-sparing effects of deflazacort [94-98].
A guideline for management of DMD patients suggests the following: (1) monitoring the presence of back pain or fractures at each visit, (2) analyzing serum calcium, phosphate, magnesium, ALP, and PTH levels at baseline, (3) baseline and subsequent annual monitoring of calcium/vitamin D intake, serum 1,25(OH)2D level, and DXA scans, and (4) lateral spine X-rays at baseline and again every 1 to 2 years thereafter if the patient is on steroids or every 2 to 3 years thereafter if not on steroids [91]. Then, if clinically significant bone fragility develops, maintenance of adequate calcium and vitamin D intake and treatment with bisphosphonate therapy are recommended. Further, DXA scans and monitoring of serum 1,25(OH)2D level, back pain occurrence, and other bone metabolism biomarkers every six months should be conducted, as well as annual spine X-rays [91].
In conclusion, many children with neurological diseases are at risk of developing osteoporosis, as they possess multiple risk factors leading to low BMD, including inactivity, decreased exposure to sunlight, poor nutrition, and use of medication or treatment that can lower BMD (e.g., AEDs, glucocorticoids, PPIs, ketogenic diet). Awareness of the increased risk of osteoporosis in these children and routine monitoring for bone health are important, as adequate management is only possible by assuring sufficient calcium and vitamin D intake and providing appropriate physiotherapy or bisphosphonate therapy.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Acknowledgements
This study was supported by a 2019 research grant from Pusan National University Yangsan Hospital.