Introduction
Autoimmune polyendocrine syndrome type 1 (APS-1), or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (OMIM 240300), is a rare autosomal recessive autoimmune disease. This disease is caused by loss-of-function mutations in the autoimmune regulator (AIRE) gene. The AIRE gene is located on chromosome 21q22.3 and encodes a 545-amino acid protein [1]. AIRE promotes ectopic expression of tissue-specific antigens in thymic medullary epithelial cells and lymph nodes, which regulate negative selection of autoreactive T effector cells [2,3]. Mutations in the AIRE gene result in decreased peripheral tissue antigen expression in the thymus. This absence ultimately leads to failure to delete the autoreactive T lymphocytes that target peripheral antigens [4].
APS-1 is clinically diagnosed by the presence of at least 2 symptoms in the classic triad, as follows: chronic mucocutaneous candidiasis, adrenal insufficiency (Addison disease), and hypoparathyroidism. The following autoimmune diseases may also occur in APS-1: type 1 diabetes mellitus, hypothyroidism, gonadal failure, autoimmune hepatitis, autoimmune gastritis, pernicious anemia, and ectodermal abnormalities (alopecia, vitiligo, keratitis, nail dystrophy, and enamel dysplasia) [5,6]. Due to its diverse symptoms and variable disease course, the clinical phenotype can differ between patients even within the same family. This phenotypic variability makes it difficult to specifically diagnose APS-1 [5,7,8].
The global prevalence of APS-1 is very low, with only 500 cases reported worldwide. There is a higher prevalence of disease in certain populations, including Iranian Jews (1:9,000) [9], Sardinia (1:14,400) [10], and Finland (1:25,000) [6]. In contrast, APS-1 is very rare in east Asia [11,12]. There are no reported data regarding its prevalence in Korea.
Here, we report a case of APS-1 in a 10-year-old Korean girl with a novel compound heterozygous mutation of the AIRE gene. Our patient’s main clinical manifestations included adrenal insufficiency and chronic mucocutaneous candidiasis.
Case report
A 10-year-old girl, born of nonconsanguineous parents, presented with poor oral intake and vomiting for 3 days. She also had general weakness, lethargy, and excessive sweating during the same period. She was born through a normal vaginal delivery at 40 weeks gestation, weighing 3.25 kg. There were no complications during pregnancy or delivery. The patient had no family history of chronic illness, including autoimmune diseases. However, she had a personal history of recurrent episodes of oral candidiasis since 6 months of age, for which she occasionally applied topical antifungal medications. She also had history of enamel dysplasia of the teeth, which was confirmed by dental examination at age 8. On physical examination, she had hyperpigmentation on the lip and fingertips. These findings had first appeared one month prior to presentation. The patient's heart rate during the initial visit was 99 beats/min, with a blood pressure of 91/68 mmHg (10th–25th percentile). She was 135 cm tall (25th–50th percentile) with a weight of 26.3 kg (10th–25th percentile), and BMI of 14.43 kg/m2 (5th–10th percentile).
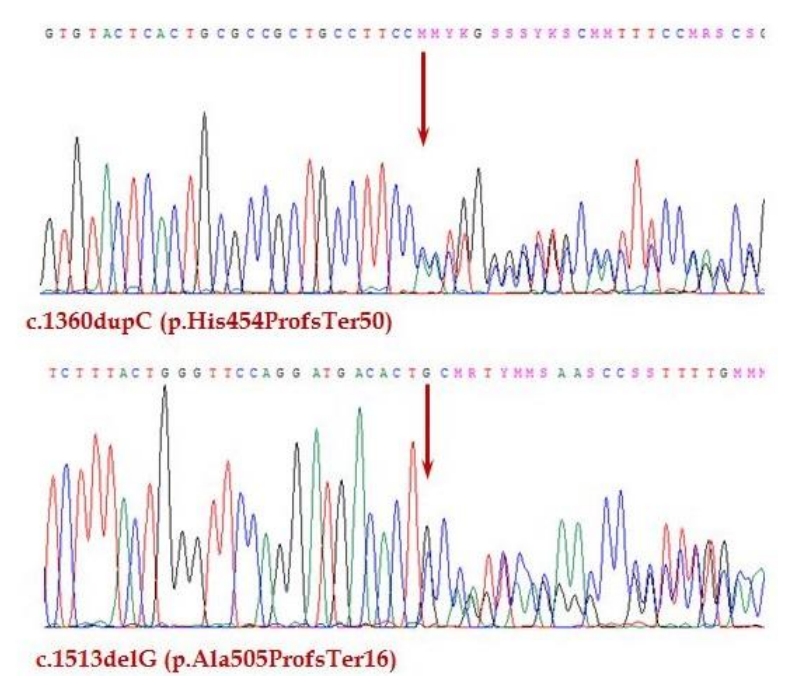
There were no abnormal findings on initial laboratory tests, including complete blood cell count and routine chemistry (Table 1). The serum sodium level was 134 mmol/L, and the potassium was 4.7 mmol/L. Hormone studies were suggestive of adrenal insufficiency, with an adrenocorticotropic hormone (ACTH) level of 1,182.0 pg/mL, and cortisol < 0.4 μg/dL. On a synthetic ACTH stimulation test, the peak cortisol level was 0.6 μg/dL. Therefore, the patient was diagnosed with primary adrenal insufficiency. Other basal hormone levels were within normal ranges (Table 2). The serum parathyroid hormone (PTH) level was 61.7 pg/mL, which is also within normal range. Oral hydrocortisone was started at a dose of 15 mg/m2. Several additional studies were performed to evaluate the etiology of adrenal insufficiency. A chromosome study showed a normal female karyotype with 46, XX. There were no remarkable findings on pelvic ultrasound or magnetic resonance imaging of the sella. A very long-chain fatty acid test for adrenoleukodystrophy was negative. The adrenal cortex antibody test was also negative. Genetic analysis did not identify any mutations in the StAR or POR genes. However, whole-exome sequencing (WES) revealed a compound heterozygous mutation in the AIRE gene. WES was performed using the TruSight One panel of 4,813 genes (Illumina, San Diego, CA, USA) for screening for pathologic mutations. One of the mutations was previously reported, while the other was novel. The previously known pathogenic variant is c.1513delG (p.Ala505ProfsTer16) [13]. The newly discovered variant is c.1360dupC (p.His454ProfsTer50) (phyloP/phastCons score 3.041/1) (http://compgen.cshl.edu/phast/index.php), which is a mutation of an evolutionarily conserved site (Fig. 1). The variant was neither found in ExAC (http://exac.broadinstitute.org/) nor 1,000 genomes (http://phase3browser.1000genomes.org). The patient's parents also underwent genetic screening. Both her father and mother were carriers of each mutation, which confirmed the etiology of APS-1 in this patient.
Discussion
APS-1 is a rare, autosomal recessive autoimmune disease that is caused by AIRE gene mutations. Today, more than 100 mutations of the AIRE gene have been reported in APS-1 patients worldwide [14]. Patients can be homozygous for both mutant alleles. Alternatively, there may be 2 independent mutations that lead to APS-1, Which was the case in our patient. The patient described here had a compound heterozygous mutation of the AIRE gene. Parental screening revealed that one mutation had originated from each parent. Mutation of c.1513delG was previously reported in 2 Japanese siblings, who were born to a Japanese mother and a Korean father [13]. In addition, a novel mutation (c.1360dupC) was found, which led to a frameshift mutation at codon 454 that created premature truncation.
APS-1 is characterized by the following 3 main clinical features: chronic mucocutaneous candidiasis, adrenal insufficiency, and hypoparathyroidism. Our patient had recurrent episodes of candidiasis since she was 6 months old. She was diagnosed with dental enamel dysplasia at 8 years old and presented with adrenal insufficiency at age 10.
Chronic mucocutaneous candidiasis is the most common initial symptom in APS-1 patients. Candida typically colonizes the oral mucosa and diaper area during infancy, and later spreads to the vulvovaginal area, intestine, and nails [15]. Chronic oral candidiasis should be treated aggressively, because uncontrolled candidiasis is associated with increased risk of esophageal strictures and squamous cell carcinoma of the oral mucosa or esophagus [16]. Therefore, it is important that these patients maintain good oral hygiene, and receive antifungal (topical) prophylaxis especially in cases of recurrent episodes of candidiasis.
Adrenal insufficiency is often diagnosed when the patient presents in adrenal crisis with rather nonspecific symptoms and signs. Patients with adrenal insufficiency usually present to the hospital complaining of general weakness, fatigue, vomiting, abdominal pain, and hyperpigmentation (similar to our case). Adrenal insufficiency is caused by a lack of adrenal hormones, including cortisol, aldosterone, and adrenal androgens. Glucocorticoid replacement therapy is required in all patients with adrenal insufficiency, and is usually treated with physiologic doses of hydrocortisone (10 to 15 mg/m2/day) [15]. It is also important to educate patients and families about managing adrenal insufficiency with "stress doses" according to the patient’s general condition.
Hypoparathyroidism is another classic manifestation of APS-1. The patient in our case did not have features of hypoparathyroidism. The incidence of hypoparathyroidism in APS-1 increases with age. One-third of APS-1 patients are diagnosed with hypoparathyroidism at age 5, two-thirds at 10 years old, and 85% at age 30 [5]. The patient in our case is 10 years old; therefore, regular follow-up with laboratory studies is necessary to monitor for signs of hypoparathyroidism, including hypocalcemia, hyperphosphatemia, and low serum PTH.
Patients with APS-1 require regular follow-up and close monitoring for the development of autoimmune diseases by pediatric endocrinologists. Autoimmune gonadal failure develops more often in female patients, who present with amenorrhea, irregular menstrual periods, and infertility [5]. Patients also must be screened regularly for type 1 diabetes mellitus, hypothyroidism, autoimmune hepatitis, and autoimmune gastritis. Diverse ectodermal dystrophies are commonly found in APS-1 patients. Enamel hypoplasia of the permanent teeth, which our patient presented with at age 8, is seen in up to 77% of APS-1 patients [6]. Other ectodermal abnormalities including alopecia, vitiligo, keratitis, and nail dystrophy, also occur frequently in APS-1.
Currently, various autoantibody tests are used in the diagnosis and follow-up of APS-1 patients. In particular, autoantibodies against interferon (α, ω) provide high sensitivity and specificity in screening for APS-1 [17]. Our patient had negative adrenal cortex antibody test results, which occurs in ~10% of autoimmune adrenal insufficiency [18]. Additional studies, including autoantibodies to interferon and organ specific antibodies, ought to be considered to monitor for additional autoimmune diseases.
The guidelines and clinical recommendations for APS-1 are insufficient given its rarity. Currently, the standard therapeutic options of APS-1 include hormone replacement and symptom control based on a clinician’s personal experience. Treatment with immunosuppressive agents, such as cyclosporine, methotrexate, and methylprednisolone is being investigated [19]. Future investigations are needed regarding genetic background, immune tolerance, and disease progress to optimize diagnosis and management of APS-1 patients.
It is important to differentially diagnose APS-1 from other autoimmune polyendocrine syndromes, such as autoimmune polyendocrine syndrome type 2 (APS-2), immune dysfunction, polyendocrinopathy, enteropathy, and X-linked (IPEX) syndrome. APS-2 is a polygenic disorder which has a complex inheritance pattern with a strong linkage to the human leukocyte antigen locus. APS-2 is defined by the presence of at least 2 of the following 3 diseases: type 1 diabetes mellitus, adrenal insufficiency, and autoimmune thyroid disease [20]. IPEX syndrome is a rare X-linked recessive disorder, which commonly presents with chronic diarrhea, type 1 diabetes mellitus, and eczematous dermatitis. It is caused by mutation in the FOXP3 gene, resulting in hyperactive T cells with both autoimmune and allergic manifestations [20]. Detailed history taking and physical exam with proper genetic studies are necessary for discriminating APS-1 from APS-2 and IPEX syndrome.
Our patient first presented with symptoms of adrenal insufficiency, and several tests were performed but the etiology was unclear until the WES analysis. Hereafter, when we encounter adrenal insufficiency patients with unknown etiology, keeping the possibility of APS-1 in mind would be helpful in early diagnosis and appropriate patient management.
In conclusion, we report a novel compound heterozygous mutation of the AIRE gene in a patient with APS-1. The patient presented with adrenal insufficiency, and had a history of chronic mucocutaneous candidiasis and dental enamel dysplasia. Further analysis including functional studies and regular follow-up are required.