Introduction
The detection of asymptomatic hyperglycemia is unusual in young children and suggests the possibility of preclinical type 1 diabetes mellitus, type 2 diabetes mellitus, and more rarely, genetic forms of diabetes [1]. Maturity Onset Diabetes of the Young (MODY) is the most common form of monogenic diabetes, resulting from ÎČ pancreatic cell and insulin secretion dysfunction [2-4]. MODY includes a clinically and genetically heterogeneous group of diabetes subtypes characterized by an autosomal dominant pattern of inheritance [2,3] with early onset of hyperglycemia in individuals, typically younger than 25 years of age [2,5,6]. MODY accounts for 2%â5% of all cases of diabetes [7]. The first gene causally implicated was the glucokinase gene (GCK) [8]. Currently, 14 genes are associated with MODY: HNF4A, GCK, HNF1A, PDX1, HNF1B, NEUROD1, KLF11, PAX4, CEL, INS, and BLK, corresponding respectively to MODY types 1 to 11, with 3 more recently discovered new genes: ABCC8, KCNJ11, and APPL1 [6,9-11]. Depending on the genetic etiology, the different subtypes present with different clinical presentations [4]. The phenotype can overlap with the classic forms of diabetes [12], and it is estimated that approximately 80% of the cases are labelled as type 1 and 2 diabetes [13]. Frequently, individuals with MODY present with one or more of the following characteristics: strong positive family history of diabetes, absence of pancreatic autoimmunity, insulin independence, evidence of endogenous production of insulin (c-peptide detection in the presence of hyperglycemia, need of low doses of insulin, or lack of ketoacidosis in the absence of insulin out of the honeymoon period), and absence of insulin resistance characteristics (absence of obesity or acanthosis nigricans) [2,4].
MODY-2 is the second most prevalent form of MODY [7]. The early onset of persistent mild fasting hyperglycemia and negative ÎČ-cell antibodies associated with a positive family history of impaired glycemia should evoke the diagnosis [6]. A definitive diagnosis is established by the identification of the mutation in the GCK gene [7].
The authors present 2 cases of early onset hyperglycemia in asymptomatic individuals with a multigenerational family history of diabetes, coexisting with dyslipidemia and obesity. A molecular study confirmed the clinical diagnosis of MODY-2.
Case reports
1. Case 1
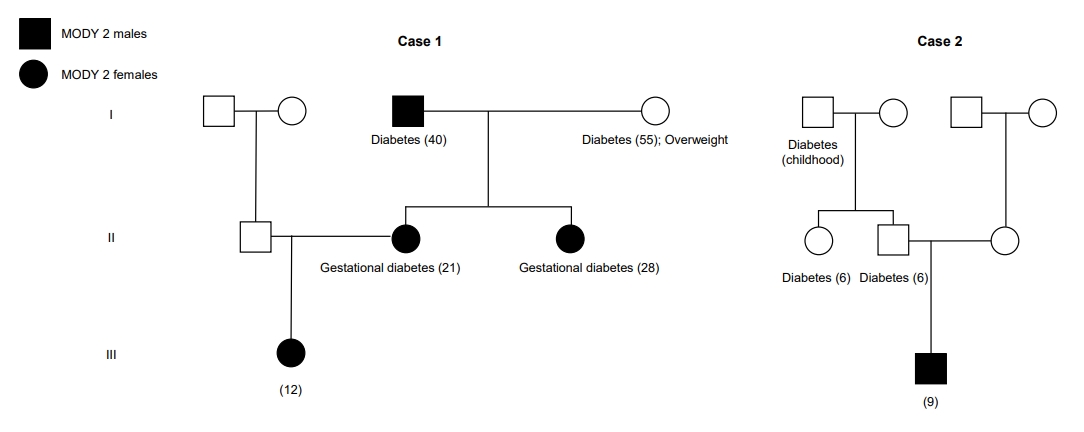
A 12-year-old girl was admitted to the Emergency Department for nonketotic fasting hyperglycemia, elevated glycated haemoglobin (HbA1c), and hypercholesterolemia detected in routine blood testing. The patient was asymptomatic and nonobese (body mass index [BMI], 19.7 kg/m2; standard deviation score [SDS], 0.37) with an unremarkable physical examination and no signs of insulin resistance.
Her family history was positive for diabetes with no apparent associated complications (Fig. 1): her mother and maternal aunt presented with gestational diabetes, her grandfather was diagnosed with diabetes and was only on dietary measures, and her grandmother was controlled with oral antidiabetic medication.
Laboratory evaluations showed a fasting glucose value of 114 mg/dL and HbA1c of 6%. Total cholesterol was 244 mg/dL (high-density lipoprotein [HDL], 52 mg/dL; low-density lipoprotein [LDL], 178 mg/dL). A standard oral glucose tolerance test with 75 g of glucose was performed with a fasting glucose of 115 mg/dL and a 2-hour glucose of 146 mg/dL. Serum islet cell autoantibodies (ICA), antibodies to insulin (IAA), glutamic acid decarboxylase (GAD), and antimicrosomal and antithyroglobulin antibodies were negative. C-peptide level was 1.3 ÎŒg/L (normal range, 1.1â5 ÎŒg/L). Follow-up evaluations were conducted and revealed fasting glucose levels between 108 and 124 mg/dL (Table 1).
The patient began treatment with gliclazide 40 mg once a day with no improvements in glycemic control, and was subsequently suspended. She was also started on simvastatin 10 mg once a day with dietetic and lifestyle measures, which were followed inconsistently.
Due to clinical and laboratory findings and the family history, the diagnosis of MODY-2 was suspected and a medical genetics consultation was performed. Sanger sequencing of the GCK gene revealed a heterozygous missense mutation c.364C>T (p.Leu122Phe) in exon 4, which was also present in the mother, maternal aunt, and grandfather.
Upon follow-up, the patient remained asymptomatic with no associated complications, presenting with an HbA1c level of 6.2% in the last laboratory evaluation at 20 years old.
2. Case 2
A 9-year-old boy was referred to the Pediatric Out-Patient Clinic for the evaluation of fasting hyperglycemia (99â126 mg/dL) and HbA1c rise (6.6%â7.6%) in postoperatory evaluations. The patient was obese with BMI 21.9 kg/m2 (SDS, 2.21) and no other findings at the physical examination, including signs of insulin resistance. The patient had a family history of diabetes in childhood (grandfather, father, and paternal aunt) (Fig. 1). However, all the affected members were prescribed dietary measures only and no complications were mentioned.
Laboratory evaluations showed an occasional glycemic value of 130 mg/dL and an HbA1c of 6.1%. A standard oral glucose tolerance test with 75 g of glucose was performed, with a fasting glucose of 117 mg/dL and a 2-hour glucose of 131 mg/dL. Serum ICA, IAA, Anti-GAD, and antimicrosomal and antithyroglobulin antibodies were negative. The C-peptide value was 2.9 ÎŒg/L (normal range, 1,1â5 ÎŒg/L). Follow-up evaluations were conducted and revealed fasting glucose levels between 117 and 135 mg/dL (Table 2).
The hypothesis of MODY-2 was suggested. A medical genetics consultation was performed and the molecular analysis of the GCK gene via Sanger sequencing revealed a heterozygous missense mutation c.698G>A (p.Cys233Tyr) in exon 7. To the best of our knowledge, this mutation was only described in the literature in another family. The genetic study was refused by the remaining family members. Based on the diagnosis of MODY-2, the proposed treatment was lifestyle modification with regular physical activity and a well-balanced diet. The patient remained overweight and exhibited height deceleration, but pathologic causes of their short stature were excluded after revealing catch-up growth suggestive of constitutional growth delay. The patient has been asymptomatic with no associated complications.
Discussion
We describe 2 pediatric cases of asymptomatic, persistent, and on-going mild hyperglycemia where the association with a multigenerational family history of early onset diabetes led us to consider MODY-2. The diagnosis was confirmed by molecular genetic testing in both cases.
In the pediatric population, MODY-2 has been reported in 40%â50% of cases of incidental or asymptomatic hyperglycemia [13]. More than 600 GCK mutations were described [2,6,7] with most of them in exons 7 and 9 [9]. The associated phenotype was similar among the various mutations and its penetrance was complete with a similar degree of hyperglycemia in the affected family members [3,10]. MODY-2 is associated with inactivating mutations of the gene that codify glucokinase (OMIM 138079), which is localized in chromosome 7p13 [10,14]. Glucokinase catalyzes glucose phosphorylation, allowing for the proper response of beta pancreatic cells and hepatocytes to the glycemic values, working like a glucose sensor [4,6]. Heterozygous loss-of-function mutations increase the glycemic threshold to insulin secretion causing mild fasting hyperglycemia (up to 120 mg/dL and a postprandial increase of 36 mg/dL up to 155 mg/dL) and elevated HbA1c (usually <8%) as well as a decreased glucose tolerance in oral glucose tolerance testing [4,8,12,13]. Individuals are usually asymptomatic and the hyperglycemia is usually detected incidentally during routine evaluations, namely during pregnancy [4,13]. However, homozygous or compound heterozygous inactivating mutations result in a more severe phenotype at birthâpermanent neonatal diabetes mellitusâleading to ketoacidosis prior to the age of 6 months requiring insulin therapy. On the other hand, heterozygous activating mutations cause hypoglycaemiaâpersistent hyperinsulinemic hypoglycemia of infancy [7,13].
Some aspects particularly suggest the diagnosis of MODY-2 as seen in our cases: fasting hyperglycemia, which is persistent (at least on 3 separate occasions) and stable over a period of months or years; HbA1c slightly above the upper limit of normal, rarely exceeding 7.5%; subtle glycemic increase 2 hours after standard oral glucose tolerance; and the presence of family members diagnosed with type 2 diabetes with no complications or symptoms [3]. MODY-2 is rarely associated with micro and macrovascular complications [2,13,15]. A study conducted in the United Kingdom reported a low prevalence of these complications in patients with a GCK mutation despite a median duration of 48.6 years of hyperglycemia [16].
The institution of dietary measures is sufficient in most cases [6,7], and pharmacologic treatment is usually not necessary given the mild hyperglycemia, the absence of long-term significant microvascular complications, and the observation that the treatment has little effect on glucose levels [2,4,10]. Patients taking insulin or oral antidiabetic medications may suspend therapy without worsening their metabolic control [12]. The only exception is during pregnancy, when insulin may be necessary to prevent fetal macrossomy [4,9,16]. Insulin secretion in the fetus in response to maternal hyperglycemia has an important role in fetal growth, and the consequences of MODY-2 on the fetus depend on whether the fetus is affected [2]. The presence of mutations in the fetus may result in a low birth weight, while the presence of mutations in the mother indirectly increases the birth weight as a result of the rise of fetal insulin secretion in response to maternal hyperglycemia. If both are carriers of the same mutation, the increase of maternal glycemia will cause appropriate fetal insulin secretion resulting in a normal birth weight [4,13]. For this reason, the identification of MODY-2 in women with gestational diabetes is important [3].
MODY diagnosis is performed through molecular genetics tests that are highly specific and sensitive, but are expensive and not easily accessible [4,12]. Confirming the diagnosis allows for the assessment of evolution and prognosis to determine the therapeutic approach and to provide genetic counselling [3,5,13]. If a mutation in the MODY gene is identified in the index patient, confirmation genetic tests should be offered to firstdegree diabetic family members. This diagnosis should not be presumed in the diabetic family members since type 1 and type 2 diabetes can coexist in MODY families [9]. Before carrying out a change in treatment, the genetic confirmation of MODY and the evaluation of the endogenous production of insulin in patients under insulin therapy is crucial. If no mutation is identified, the MODY diagnosis is unlikely, but cannot be excluded, due to the possibility of rare or still unidentified mutations [9]. Nonaffected family members can be initially scanned through fasting glucose testing, and if high, the diagnosis should then be confirmed through genetic testing [3,9].
In both presented cases, MODY-2 diagnosis was evoked by the presence of early onset, nonprogressive, and persistent fasting hyperglycemia detected in routine laboratory evaluations in asymptomatic individuals, as well as the absence of anti-ÎČ pancreatic cell antibodies and compatible family histories. However, the presence of dyslipidemia could initially suggest the diagnosis of type 2 diabetes in the first case. Although dyslipidemia may occur in MODY-2 patients, patients with GCK mutations usually exhibit low serum levels of fatty acids and triglycerides compared to healthy individuals because reduced GCK activity interferes with hepatic fatty acid and triglyceride production. MODY-2 patients seem to show a protective profile of HDL cholesterol [15]. Still, the absence of obesity and signs of insulin resistance, the presence of mild fasting hyperglycemia and subtle glycemic increase in the oral glucose tolerance test, and the lack of response to oral antidiabetic drugs, combined with the family history, strongly suggested MODY-2.
On the other hand, the presence of obesity in the second case, not commonly associated with MODY-2 patients [15], could lead to doubts about the diagnosis. However, the lack of other clinical findings, the laboratory evolution, and family history were crucial in the differential diagnosis. Molecular testing confirmed the diagnosis of MODY-2.
In both cases, the identified variants were checked against relevant databases and were considered pathogenic in the Human Gene Mutation Database and likely pathogenic in Varsome with no report in the Clinvar database [17-20]. In the second case, to our knowledge, the identified GCK variant has only been described as a novel mutation in another family in New Zealand that was strongly affected by both MODY and classic autoimmune mediated diabetes, including one individual with latent autoimmune diabetes [5]. In our case, although an early onset of diabetes was reported in 2 family members, it was not described as antibody positive type 1 diabetes and no treatment measures were necessary with no associated complications.
These findings indicate that the phenotype heterogeneity was associated with GCK mutations, sometimes overlapping with other types of diabetes.
In conclusion, MODY-2 should be considered in patients with asymptomatic hyperglycemia and a high level of suspicion must be exhibited in the presence of compatible clinical findings and family history. These cases highlight the challenges and the required level of suspicion of a diagnosis that has significant implications, not only in appropriate management and clinical outcomes, but also in genetic counselling and the subsequent identification of affected family members.