Introduction
Growth hormone transduction defect (GHTD) is a newly recognized growth disorder, characterized by severe short stature as a result of defective GH signaling. The biochemical findings of this disorder include low insulin-like growth factor-1 (IGF1) concentrations, normal GH response to provocative tests, normal 24-hour GH concentrations and increased expression of IGF1 during the IGF1 generation test. Regarding the molecular mechanisms involved, it is characterized by impaired phosphorylation of signal transducer and activator of transcription-3 (STAT3), baseline over-expression of the GH negative regulator, cytokine inducible SH2 containing protein (CIS), and overexpression of the cell cycle inhibitor, p21/CIP1/WAF1 (p21). Fibroblasts from GHTD patients showed significantly decreased cell proliferation and a cell cycle pause mostly in the G0 phase, which normalized after induction with an increased GH dose, as opposed to normal fibroblasts [1]. Patients with GHTD exhibit "catch-up" growth after exogenous recombinant human GH (rhGH) treatment and manage to achieve a normal final adult height [2].
Our previous studies have shown that impaired GH signaling is due to overexpression of ubiquitinated CIS that causes rapid and premature translocation of the GHR to the proteasome for degradation, through its action as an E3 ubiquitin ligase [2]. Induction of GHTD fibroblasts with 1,000-╬╝g/L human GH (hGH) (5 times the amount of hGH needed in control fibroblasts) restored the GHR to the plasma membrane and resulted in STAT3 phosphorylation and normal GH signaling [2]. In addition, one of our previous studies suggests that the EGF pathway is involved in the restoration of the GH pathway after induction with the higher dose of hGH [3].
Fibroblasts of GHTD children show a cell-cycle arrest at the G0/G1 phase in association with p21 nuclear overexpression [1]. p21, a downstream target gene of p53, encodes a protein that regulates cell cycle and DNA synthesis. It is an inhibitor of the cyclin-dependent kinases (CDK), which are responsible for progression through the cell cycle. It binds to G1 CDKs and acts as a regulator of the cell cycle progression to S phase. It also binds to the proliferating-cell nuclear antigen (PCNA), a DNA polymerase ╬┤ processivity factor which directly interferes with DNA replication. Therefore, the progression to S phase is prevented in 2 ways: by inhibition of G1 CDKs and inhibition of PCNA [4,5]. p21 has both negative and positive functions in the cell cycle, mediating either suppression or promotion of cell proliferation depending on its intracellular localization. More specifically, when p21 is located in the nucleus, it inhibits the activity of cyclin-dependent kinases CDK1 and CDK2 and blocks the transition from the G1 phase into the S phase or from the G2 phase into mitosis. Cell cycle arrest allows cells to repair DNA damage before cell division starts again [6]. However, when p21 is located in the cytoplasm, it has an antiapoptotic action by binding to and inhibiting caspase 3, as well as the apoptotic kinases, apoptosis signal-regulating kinase 1 and c-Jun N-terminal kinase [7-10].
Futhermore, p21 has an important biological role in the negative feedback regulation of cytokine signaling pathways. It has also been described that p21 participates in STAT3 pathway inhibition by interacting with STAT3 proteins, blocking their transcriptional activity and preventing the binding of the CBP coactivator. Conversely, activation of STAT proteins leads to transcriptional activation of the p21 gene and induces its protein synthesis [11].
Therefore, we investigated the role of p21 in the negative regulation of the GH/GHR and EGF/EGFR pathways in GHTD.
Materials and methods
Fibroblast cultures were developed from gingival biopsies of one prepubertal GHTD patient (P) and one prepubertal age-matched control child (C). The GHTD child was 8 years and 6 months of age, with a height standard deviation score (SDS) of -2.42 and bone age retardation SDS of > -3.0. The control child (C) had normal stature. Both children were recruited from the outpatient clinic of the Division of Pediatric Endocrinology and Diabetes of the University Hospital of Patras, Greece. The study was approved by the Ethics Committee of the University Hospital of Patras in Greece (A2╬▓/╬ō.╬Ā.:82556/21-11-2016). After informed parental consent, the child with GHTD was treated with rhGH and reached an adult height of 174 cm (SDS, -0.44).
1. Cell cultures
Fibroblast cultures were established from gingival biopsies obtained from the patient and the control after informed parental consent and children's assent. The tissue pieces (0.5ŌĆō1.0 mm3; 2ŌĆō3 pieces per dish) were plated onto 60-mm2 culture dishes in Dulbecco's Modified Eagle's Medium (DMEM), supplemented 2-mM L-glutamine (Gibco, Invitrogen, Life technologies, Carlsbad, CA, USA), 10% fetal bovine serum (FBS, Gibco, Invitrogen, Life technologies), 50-IU/mL penicillin and 50-╬╝g/mL streptomycin (Gibco, Invitrogen, Life technologies), and incubated at 37Ōäā in 5% CO2 atmosphere. Cultures were replenished with fresh medium every 3 days and then subcultivated in a 1:3 split ratio upon reaching confluency by using a trypsinŌüäEDTA solution (Gibco, Invitrogen, Life technologies).
2. GH and EGF inductions
The patient and control fibroblasts were cultured at 80% confluency, washed twice with phosphate buffered saline, maintained for a 24-hour period in serum-free DMEM and then treated with 2 different concentrations of hGH (Humatrope, Lilly, Indianapolis, IN, USA): 200 ╬╝g/L (GH200) and 1,000 ╬╝g/L (GH1000) or with EGF (human recombinant EGF, Millipore, Billerica, MA, USA) 50 ng/mL (EGF).
3. Silence CIS (short interference RNA CIS/siCIS)
The cellular localization of p21 was studied at baseline and after induction with GH200, either with or without siRNA CIS.
Silence CIS (siCIS), (Santa Cruz Biotechnology Inc., Dallas, TX, USA), was performed according to the manufacturerŌĆÖs instructions. A total of 2├Ś105 cells/well were cultured in 2-mL antibiotic-free DMEM supplemented with FBS until the cells were 60%ŌĆō80 % confluent. For each transfection, 1 ╬╝g of siCIS was diluted into 100-╬╝L siRNA Transfection Medium (sc-36868), Solution A, and 6 ╬╝L of siCIS Transfection Reagent (sc-29528) into 100-╬╝L siCIS Transfection Medium (sc-36868), solution B. Solution A was added to solution B and the mixture was incubated for 45 minutes at room temperature.
The fibroblasts were washed once with 2 mL of siCIS Transfection Medium (sc-36868). The medium was aspirated. For each transfection, 0.8 mL of siCIS Transfection Medium was added to each tube containing solution A and solution B and the mixture was applied onto the washed cells. The cells were then incubated for 7 hours at 37Ōäā in a CO2 incubator. Subsequently, 1 mL of normal growth medium containing 2 times the normal serum and antibiotics concentration was added to the transfection mixture and the cells were incubated for an additional 24 hours. The medium was then aspirated and replaced with fresh 1├ŚDMEM for 24 hours. The cells were lysed for Western Immunoblotting.
4. Western immunoblotting
The protein expression of p21 was studied by Western immunoblotting, as follows: (1) at the basal state and after induction of the childrenŌĆÖs fibroblasts with 200-ng/mL hGH (GH200), either with or without siCIS and (2) at the basal state and after induction of the fibroblasts for 15 minutes with GH200, 1,000 ng/mL (GH1000) or EGF. Western blotting was performed as previously described [3]. p21 antibody (Novus Biologicals, NBP1-04753) was used at the indicated concentrations: 1:500.
The cell lysates were diluted in homemade sodium dodecyl sulfate sample buffer that was supplemented with ╬▓-Merkaptoethanol (Sigma Aldrich, St. Louis, MO, USA) and denatured. The mean protein concentration, before sodium dodecyl sulfate was added, was 1 ╬╝g/╬╝L (50 ╬╝g/50 ╬╝L) of the lysate. Coomassie blue staining of the gel was used as a loading control. Then, the cell lysates were loaded into wells in a sodium dodecyl sulfate-polyacrylamide gel (10%). Fifty microliter of each sample were loaded into each lane. A prestained standard (Benchmark Prestained Protein Ladder, Invitrogen, Life technologies) was used as a molecular weight marker. Voltage was applied along the gel and the proteins were separated dependent on their size. Following electrophoresis, the proteins were transferred to a nitrocellulose transfer membrane (Whatman, Protran, Sigma Aldrich), which was then incubated with the primary antibody for 12 hours and the appropriate second antibody for an additional 1 hour. Immunoreactive protein was visualized by chemiluminescence after the membranes were exposed to ECL (H2O2 and luminal, GE Healthcare Biosciences AB, Uppsala, Sweden) and developed on photographic films. Autoradiograms were scanned and the bands corresponding to p21 were analyzed by densitometry. The protein expression of p21 was compared to the protein expression of ╬▓-tubulin, by using the program Scion image (version 4.0.3.2; Scion Corp., Chicago, IL, USA).
5. Immunofluorescence
The cellular localization of p21 was studied with immunofluoresence. For the first part of the experiments, cells at the basal state or after induction with 200 ╬╝g/L, were treated either with siRNA CIS or without. In the second part of the experiments, after induction of the cells with hGH (200 ╬╝g/L or 1,000 ╬╝g/L) or EGF (50 ng/mL) or without induction, the cells were washed twice with phosphate buffered saline. Cells were fixed and permeabilized. The fixed cells were blocked for 1 hour with 1% bovine serum albumin. The primary antibody was applied at an appropriate dilution, time and temperature (p21, 1:50, overnight at 4Ōäā), (Santa Cruz Biotechnology Inc.; sc-817). After the incubation with the primary antibody, the cover slips were washed three times with 1% bovine serum albumin followed by a 1-hour incubation period with the secondary antibodies (FITC goat anti-mouse IgG-FITC, sc-2010, 1:100, Santa Cruz Biotechnology Inc.). The images were acquired on a Nikon Eclipse TE 2000-U.
Results
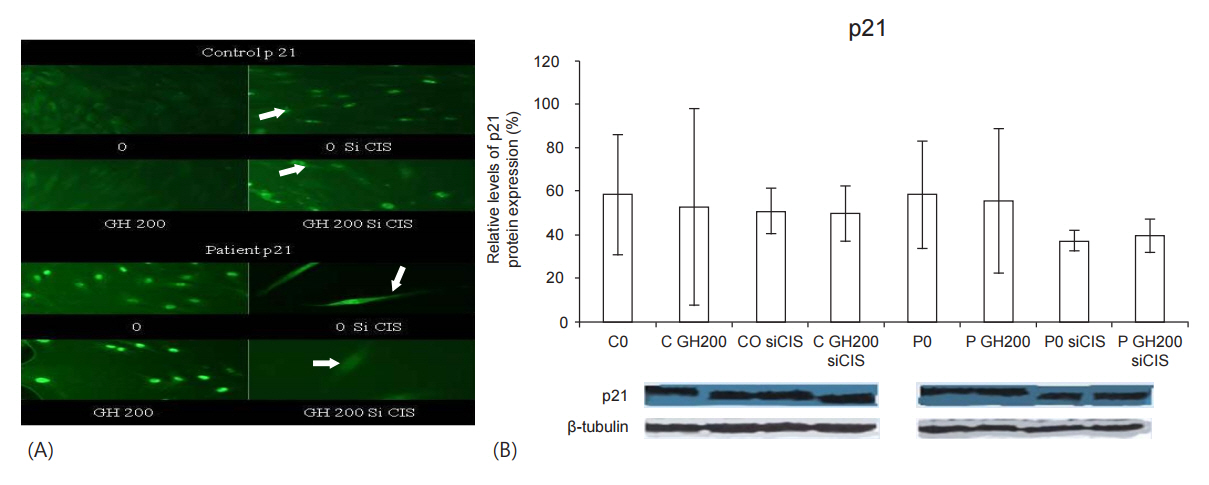
After silencing of the CIS gene (siCIS), either with or without induction of the fibroblasts with GH200: (1) in the control, the nuclear localization of p21 was intensified compared to that seen without siCIS; (2) in the GHTD patient, p21 was translocated from the nucleus to the cytoplasm (Fig. 1A); (3) the protein expression of p21 was reduced in the control and even significantly more in the patient (Fig. 1B).
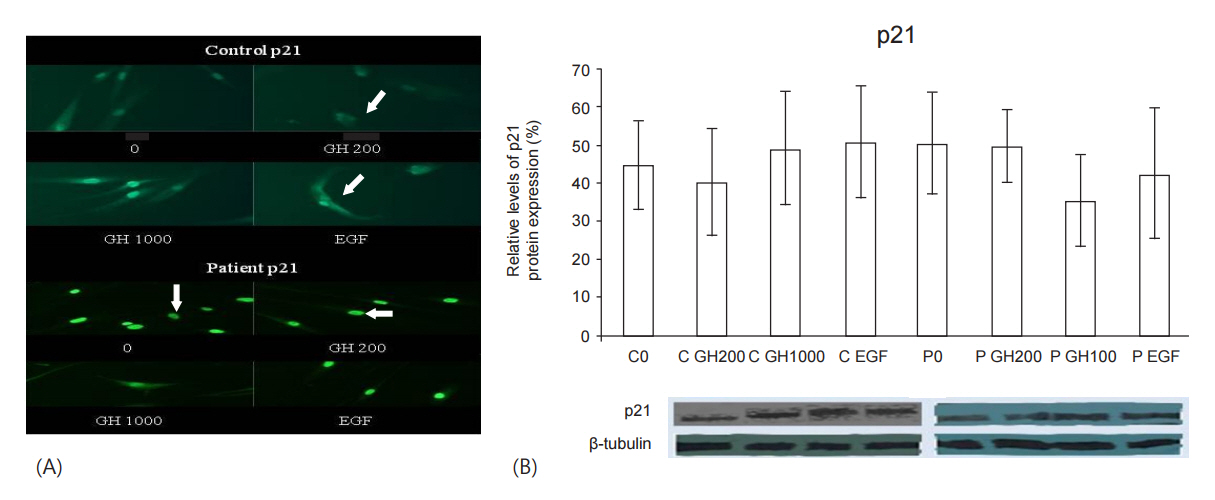
After the inductions of successful GH signaling (GH200 for the control and GH1000 for the patient, as shown from our previous studies [1]): (1) in the control, nuclear localization of p21 was decreased in favor of cytoplasmic localization (Fig. 2AŌĆōarrows) and its protein expression was decreased compared to the baseline (Fig. 2B), (2) in the GHTD patient, p21 was translocated to the cytoplasm (Fig. 2AŌĆōarrows) and its protein expression was greatly reduced (Fig. 2B).
After induction with EGF, where the protein and membrane expression of EGFR and pEGFR are increased [3]: (1) in the control, p21 was located both in the cytoplasm and in the nucleus, whereas in the GHTD patient, p21 was sustained in the nucleus as observed at baseline (Fig. 2AŌĆōarrows). (2) in the control, the protein expression of p21 was increased compared to baseline, whereas in the patient it was decreased (Fig. 2B).
Discussion
GHTD is a growth disorder which has given greater insight into factors influencing GH signaling under normal and abnormal conditions. Thus far, our research into the molecular mechanisms of this disorder has enhanced a further understanding of the interrelationships between the GH and EGF pathways, as well as the GH and cellular regulators, CIS and p21 [1-3].
Our current study suggests that there is a positive correlation between CIS and p21 protein expression, which seems to be more prominent in the GHTD fibroblasts. This can be seen by the reduction of p21 protein expression after siCIS in the control and, more evidently, in the GHTD fibroblasts. Of interest, though, is the differential impact of siCIS on the cellular localization of p21 in the respective fibroblasts. The increased nuclear p21 localization (which favors cell cycle arrest) in the control fibroblasts may suggest that the silencing of the GH inhibitor, CIS, possibly advocates p21's antiproliferative action in order to control aberrant cellular growth. On the other hand, the increased cytoplasmic localization of p21 in the GHTD fibroblasts after siCIS, which favors p21's antiapoptotic action, may play a role in enhancing cellular growth. These results are in agreement with our previous findings [2]. Another possible indication of the positive correlation between p21 and CIS is the reduction in the protein expression of p21 after hGH induction when GH signaling is successfully activated in the control and the GHTD fibroblasts (control, GH200; patient, GH1000), where, according to our previous studies, CIS is also reduced. It is of interest, though, that after these hGH inductions, the cytoplasmic localization of p21 is increased in both the control and GHTD fibroblasts, possibly favoring p21's antiapoptotic role in both cases.
It is well known that the expression of p21 is negatively regulated at the transcription level by Telomeric Repeat Binding Factor 2 (TRF2) [12]. A repressor complex is engaged to the p21 promoter in a TRF2-dependent fashion, inhibiting the transcription of the p21 gene [13]. TRF2 is degraded by ubiquitinmediated proteolysis in mammalian cells [14,15]. Therefore, the results of our current study suggest that the positive correlation observed between CIS, an E3 ubiquitin ligase, and p21/CIP1/WAF1, may be explained by the ubiquitin-mediated proteolysis of TRF2 in the proteasome, possibly followed by enhanced transcription of p21. It has also been documented that activation of EGFR is followed by activation of human telomerase reverse transcriptase (hTERT) mRNA expression. TRF2 is mechanistically involved in the transcriptional interaction of the hTERT promoter. On the other hand, expression of p21 attenuates hTERT promoter activity, mediating attenuation of telomerase activity and cell proliferation [12,16,17]. Further studies are necessary for the role of these interactions to be clarified.
It has been documented that in intestinal epithelial cells EGFR stimulation increases p21 expression and its cytoplasmic localization and facilitates the assembly of Cyclin D1/Cdk4 complexes, promoting cell cycle progression and cell proliferation [18]. This is mediated by the activation of PI3K/AKT [19]. Similarly, in our current study we noted an increased protein expression and cytoplasmic localization of p21 in the control fibroblasts after induction with EGF, suggesting p21's anti-apoptotic action in this case. On the contrary, though, in the GHTD fibroblasts, after EGF induction, the protein expression of p21 was decreased and it was localized in the nucleus, favoring its action as a cell cycle inhibitor and suggesting its participation in the negative regulation of the EGF/EGFR pathway. Therefore, a decrease in p21 could possibly be a prerequisite for successful activation of the EGF/EGFR pathway in GHTD.
It has been shown in previous studies that epidermal growth factor (EGF) was able to induce CIS gene expression moderately in mammary epithelial and mainly liver mice cells [20]. We have previously shown that after EGF induction of GHTD fibroblasts, CIS protein expression was reduced, suggesting that CIS may play a role in the inhibition of the EGF/EGFR pathway in this disorder [3]. This again suggests a positive relationship between CIS and p21, but only in the GHTD patient in the case of the EGF/EGFR pathway.
This is a unique study which further elucidates the molecular mechanisms of GHTD. The results of these molecular analyses are important as they enhance our understanding of the complexity and diversity of the molecular interactions between cellular signal cascades, which regulate both cell proliferation and the negative regulation of the cell cycle. This is of great value for the understanding of pathological processes such as growth deficiency, degenerative diseases and neoplastic processes. This study, as a qualitative one, has some limitations that should be considered. The molecular studies were performed in a model of one patient and one control, which limits the generalizability of the findings, however, a large number of molecular analyses were performed and the results were verified.
In conclusion, it is possible that overexpression of p21 may not only be involved in the negative regulation of the GH/GHR pathway in GHTD, but also in that of the EGF/EGFR pathway. Hence, our present study provides further information regarding the possible pathogenesis of GHTD in relation to p21. More studies are needed to further elucidate the mechanisms involved in GHTD, which may offer further insight into additional molecular mechanisms involved in children with growth impairment.