Introduction
Neonatal diabetes mellitus (NDM) is characterized by the onset of diabetes before 6 months of age. NDM has an almost monogenic etiology and is considered to be a rare condition, with a prevalence of 1 in 100,000ŌĆō300,000 live births [1]. NDM includes permanent neonatal diabetes mellitus (PNDM), which requires insulin or oral sulfonylurea treatment for life, and transient NDM, which usually resolves at a median age of 12 weeks, but approximately 50% of cases relapse in adolescence or young adulthood [1]. PNDM is most commonly a result of heterozygous mutations of the genes KCNJ11 and ABCC8 encoding the ATP-sensitive potassium channel (KATP) [1,2]. Approximately half of the cases are caused by a mutation in the KCNJ11 gene [1,3]. KCNJ11 mutations result in decreased sensitivity of the Kir6.2 subunits to ATP, with channel closure failure and consequently insufficient insulin release from the ╬▓-cells [2]. Developmental delay, epilepsy, and neonatal diabetes (DEND) syndrome are described in some patients with KCNJ11 mutations [1]. In most patients with KATP-related diabetes, oral sulfonylurea therapy promotes insulin secretion through the ATP-independent closure of overly active mutated channels [1-3]. Sulfonylureas represent a suitable therapy for patients with KCNJ11 mutations, instead of insulin therapy. However, not all patients are successfully treated with sulfonylurea [3].
Only a few cases of PNDM have been reported in Korea [4-7]. Of the five cases, patients with p.K170R, p.V59M, and p.H186D mutations showed successful treatment with sulfonylurea [4,6]. Only patients with p.V59M mutation showed moderate developmental delay [6]. Two cases of patients with p.R201H mutation have been reported [5,7]. One case was unsuccessfully treated with a sulfonylurea with no neurologic symptoms [7], and another case showed DEND syndrome with a successful switch from insulin to sulfonylurea therapy [5]. Herein, we report a case of PNDM caused by a heterozygous p.G53D mutation in the KCNJ11 gene with successful glycemic control by initiating sulfonylurea treatment at 3 months of age.
Case report
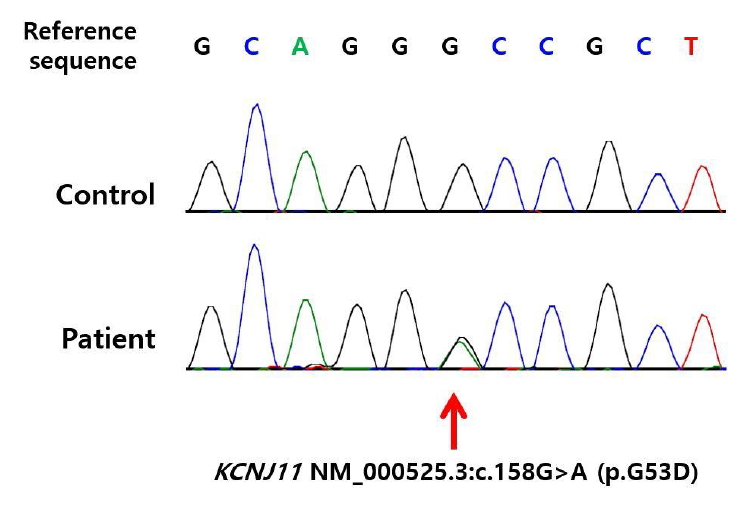
A 53-day-old girl was hospitalized owing to diabetic ketoacidosis (DKA) and a generalized tonic-clonic seizure in March 2017. The patient was born at 38 weeks of gestation and demonstrated intrauterine growth retardation. She was delivered via normal spontaneous vaginal delivery and weighed 2,520 g (standard deviation score [SDS], -1.89) at birth. She was the first child and had no siblings. Her family had no history of diabetes. Laboratory results at diagnosis were as follows: blood glucose, 702 mg/dL; pH, 6.979; HCO3-, 4.5 mmHg; base excess, -25.7 mmol/L; total CO2, 5 mmol/L; sodium, 141 mEq/L; corrected sodium, 149 mEq/L; potassium, 6.5 mEq/L; chloride, 105 mEq/L; urine glucose, 3+; urine ketone, 3+; glycated hemoglobin (HbA1c), 7.8%; C-peptide, <0.01 ng/mL; insulin 6.6 ╬╝IU/mL; insulin antibody 6.6% (0.0ŌĆō7.0); glutamic acid decarboxylase II antibody, 0.59 U/mL (0.0ŌĆō1.0); and islet cell antibody, negative. The patient recovered from DKA, and the continuous insulin treatment was subsequently discontinued. Instead, the patient had an injection of glargine (Lantus) once daily and regular insulin twice daily. Her insulin requirement gradually increased to 0.9 U/kg/day, and the seizure was no longer observed. No abnormal findings were observed on both the electroencephalogram and brain magnetic resonance image. She was hospitalized for a total of 14 days. Direct sequencing of the KCNJ11 and ABCC8 genes using genomic DNA extracted from peripheral lymphocytes was conducted during the hospitalization; her parents were not subjected to genetic testing. Six weeks later, direct sequencing of the KCNJ11 gene, encoding the Kir6.2 subunit of the KATP channel, confirmed the presence of a heterozygous mutation, KCNJ11:c.158G>A (p.G53D) (Fig. 1). ABCC8 mutation was not detected. By then, the patient was 3 months of age, and insulin was administered at 1 U/kg/day. She could lift her head and did not demonstrate signs of developmental delay. We immediately started sulfonylurea (glimepiride) at 0.3 mg/kg/day and gradually tapered the insulin dosage. Five weeks later, insulin was discontinued, and the dosage of glimepiride was adjusted to 0.75 mg/kg/day. The patient is now 11 months of age, and her weight is 9.7 kg (SDS, 0.07). She can sit without help, pull herself to the standing position, and respond well to simple verbal requests. She is also able to pronounce simple words like "mama." She maintains excellent blood glucose control without hypoglycemic events, with an HbA1c level of 6.3%. Six months after the initiation of glimepiride therapy, her serum C-peptide level was 2.53 ng/mL, which was higher than the initial level of <0.01 ng/mL. Her current glimepiride dose is 0.6 mg/kg/day. Thus far, no further episodes of seizure have been reported.
Discussion
In this case, a 53-day-old girl was diagnosed with NDM due to a p.G53D mutation in KCNJ11 via early genetic testing upon hospital admission owing to DKA. Early switching from insulin to sulfonylurea treatment brought about successful blood glucose control.
NDM is a rare disease and is characterized by insulin-dependent persistent hyperglycemia that develops over the first 6 months of life [1]. Diabetes mellitus that begins before 6 months of age is unlikely to be caused by an autoimmune disease, but instead by monogenic inheritance. Mutations in the KCNJ11 and ABCC8 genes account for 30%ŌłÆ50% of cases [1,3]. KCNJ11 gene mutations cause failure of ╬▓-cell KATP channel closure in response to intracellular ATP elevation, resulting in impaired insulin release. Sulfonylureas close the KATP channel in an ATP-independent manner [1,3] and have been reported to be a suitable treatment for patients with diabetes due to KCNJ11 mutations [2,3,5,8-10].
Successful change from insulin therapy to sulfonylurea is known to be affected by the severity of clinical symptoms and duration of the disease. Pearson et al. [3] reported that 90% of patients (44 of 49) successfully switched from insulin to oral sulfonylureas and 5 patients (10%) unsuccessfully switched. The success rate is lower in patients with DEND syndromes. Four of 5 patients (80%) who were unable to switch to treatment with sulfonylureas had neurologic features. In contrast, 6 patients (14%) whose treatment was successfully switched to sulfonylureas had neurologic features. The median age of initiation of sulfonylurea treatment in the successful group was 6 years (interquartile range, 3ŌĆō12 years); this was younger than the unsuccessful group with median age of 18 years (interquartile range, 6ŌĆō35 years). Patients with p.R201H mutations have undergone both successful switching and unsuccessful switching from insulin to sulfonylurea [3,7]. p.R201H mutations in KCJN11 accompanying older age at initiation of sulfonylurea treatment are well-known factors influencing unsuccessful switching. In the case of unsuccessful switching from insulin to sulfonylurea, the patient with p.R201H mutations was 18 years old at the time of switching [7]. Other studies have reported that early initiation of sulfonylurea treatment has a beneficial effect on remission of neonatal diabetes [11]. In addition, in a murine model study, early sulfonylurea treatment leads to permanent remission of NDM [12]. This suggests that sulfonylurea treatment as early as possible may help in the successful switching insulin to sulfonylurea.
Treatment of DEND syndrome using sulfonylurea affects the prognosis. Previous clinical reports have shown that patients with DEND syndrome demonstrate improved neurological symptoms when a sulfonylurea is administered at an early stage of the diagnosis or adulthood [8,12,13]. There has been a report of a 17.9-year-old patient with DEND syndrome for whom sulfonylurea treatment was initiated and who achieved excellent glycemic control, but psychomotor retardation did not improve [5]. As the brain develops continuously through infancy and beyond, the early administration of sulfonylureas may offer significant benefits to the neurodevelopmental outcomes [14]. Therefore, the importance of early administration of sulfonylureas is emphasized for a better neurodevelopmental outcome as well as for stable blood glucose control.
To our knowledge, there were no serious adverse effects reported in the treatment of sulfonylurea in NDM. Most cases reported on the efficacy of glimepiride as a treatment for KCNJ11 mutation, and there were some reports of the effects of gliclazide and glibenclamide [2,3,5,8-10]. All these drugs are second-generation drugs and are generally known to have an equivalent effect in lowering blood glucose concentration [15]. Glibenclamide has been associated with an increased risk of hypoglycemia and cardiovascular-related mortality in adult type 2 diabetes mellitus patients [16]. Adverse effects due to glibenclamide administration in NDM patients have been reported with transient diarrhea [17] and tooth discoloration [18]. Significant side effects of administration of glibenclamide have not been reported in the treatment of NDM, and it is generally regarded as a safe treatment. According to a case report of glimepiride administration in NDM patients, a patient who was initially misdiagnosed with type 1 diabetes mellitus successfully converted to sulfonylurea treatment at 37 years of age and was given glimepiride due to the possible side effects of glibenclamide described above [9].
A total of 5 cases of NDM due to mutation of the KCNJ11 gene have been reported in Korea (Table 1) [4-7]. The mean age at initiation of sulfonylurea treatment was 10.2┬▒7.1 years (3.8ŌĆō18.0 years), and failure to switch from insulin therapy to sulfonylurea therapy was reported in one case. In our patient, p.G53D mutation was found, and the patient showed successful conversion from insulin to sulfonylurea treatment. Glimepiride was administered instead of glibenclamide. Our case showed that glimepiride is effective in achieving successful blood glucose control and causes no side effects for patients with NDM due to p.G53D mutation. The p.G53D mutation in KCNJ11 has previously been reported in a patient with DEND syndrome [8,19], which comprises developmental delay, seizures during infancy, and improved motor features after switching from insulin to sulfonylurea treatment [8]. Given the report of patients with this case and previously reported p.G53D mutation in patients with DEND syndrome [8], patients with p.G53D mutation can also be considered to successfully convert from insulin to sulfonylurea treatment. The patient in the present case could not be diagnosed with DEND syndrome because developmental delay had not been confirmed yet. There is no developmental delay so far. However, considering that the p.G53D mutation is associated with DEND syndrome [8,19], she will require continuous monitoring of her developmental status.
In conclusion, genetic testing is an indispensable diagnostic tool for diabetes mellitus patients under 6 months of age because it can provide an accurate diagnosis and effective treatment, which improves the prognosis. Additionally, glimepiride administration in NDM associated with p.G53D mutation can effectively control blood glucose.