Introduction
Pituitary stalk interruption syndrome (PSIS) involves the occurrence of a thin or absent pituitary stalk, hypoplasia of the adenohypophysis, and ectopic neurohypophysis [1]. Diagnosis of this disease is confirmed using magnetic resonance imaging (MRI). PSIS was first reported in 1987, based on MRI results [1]. The incidence of this syndrome is approximately 0.5 per 100,000 births. Patients with PSIS patients have pituitary hormone deficiencies with variable timings of onset [2]. Hence, manifestations of PSIS have a wide spectrum of clinical phenotypes. For example, Olszewska et al. [3] published a case report of severe panhypopituitarism with PSIS in a newborn (4-day-old male) delivered by a mother with Turner Syndrome. Marmouch et al. [4] reported a case of late PSIS onset in which a 17-year-old female patient presented primary amenorrhea and impuberism. Symptoms of PSIS can be overlooked during the neonatal period and infancy, therefore, diagnosis can be delayed.
Of the 2 posterior pituitary hormones, antidiuretic hormone (ADH) deficiency leads to diabetes insipidus. The major symptoms are polyuria, polydipsia, and nocturia. Under this condition, hypernatremia as well as extreme thirst can easily occur. However, diabetes insipidus may be masked when accompanied by an anterior pituitary hormone deficiency, such as ACTH deficiency, in which ADH is enhanced due to a cortisol deficiency [5]. Diederich et al. [6] described the patientŌĆÖs illness as being similar to the syndrome of inappropriate antidiuretic hormone secretion (SIADH). To best of our knowledge, as of yet, there has been no report of severe hyponatremia associated with hypopituitarism in Korean children. Here, we report a patient with congenital hypopituitarism presenting with severe hyponatremia as the first symptom.
Case report
At the age of 24 years, the patient was admitted to a university hospital with right upper quadrant pain and fever. He was suspected of having cholecystitis and was treated with intravenous (IV) antibiotics and NPO (nil per os). His symptoms improved after 2 days, but he was transferred to nephrology clinic of Kyungpook National University Hospital for uncorrected hyponatremia. After initial management, he was also referred to pediatric endocrinology clinic for short stature, hypogonadism, and hypothyroidism.
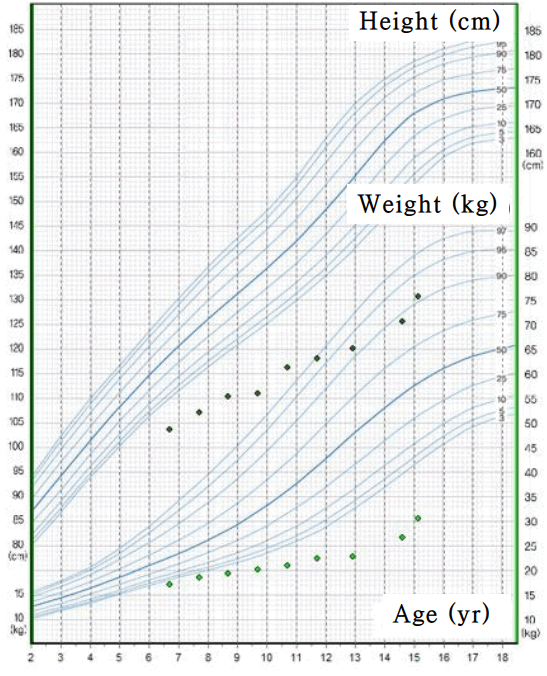
On admission, the patientŌĆÖs weight and height were 36.6 kg (<3rd percentile) and 146 cm (<3rd percentile), respectively, and he had a blood pressure of 108/60 mmHg. There was no evidence of dehydration, and his creatinine and uric acid levels were also within normal range. He showed no development of secondary sex characteristics, such as enlargement of testis and penis, or deepening of the voice. The grade of pubic hair was stage I on sexual maturity ratings, and his testicular volume was markedly decreased to 2ŌĆō3 mL. When we reviewed past medical history during childhood, we could see our patient already had markedly short stature and growth retardation. (Fig. 1). Laboratory examinations revealed profound hyponatremia at 114 mmol/L, and a normal level of potassium at 4.0 mmol/L. At that time, urinary sodium concentration was 87 mmol/L. Unfortunately, his serum osmolarity and urine osmolarity were not measured upon admission. We checked serum/urine osmolarity and plasma arginine vasopressin (AVP). Laboratory findings showed that serum osmolarity and urine osmolarity levels were 285 mOsm/kg and 509 mOsm/kg, respectively; urine osmolarity was markedly increased compared to serum osmolarity. Plasma AVP level was 8.96 pg/mL (normal, <2.5 pg/mL), showing a sustained elevation despite several days of hydrocortisone treatment due to low cortisol level.
He had been born without perinatal complication at 40 weeks of gestation and had a birth weight of 3,300 g by normal vaginal delivery. His parents denied the patientŌĆÖs hospitalization for hypoglycemia or seizure and micropenis during infant.
At the age of 14 years, this patient had been admitted for meningitis 10 years prior to his most recent admission. At that time, his laboratory results showed severe hyponatremia (115 mmol/L) without hyperkalemia (potassium at 3.5 mmol/L) and he suffered a generalized tonic-clonic type seizure and was treated with mechanical ventilation for 6 days. Serum osmolarity and urine osmolarity levels were 249 mOsm/kg and 352 mOsm/kg, respectively, urinary sodium concentration was 103 mmol/L. His hyponatremia was regarded as SIADH due to meningitis. He was treated with IV antibiotics, fluid restriction for SIADH and IV hydrocortisone for persistent hypotension. He was also diagnosed with hypothyroidism with free T4 levels at 0.10 ng/dL (normal, 0.8ŌĆō1.0 ng/dL) and thyroid stimulating hormone (TSH) levels at 3.20 ╬╝IU/mL (normal, 0.3ŌĆō4.0 ╬╝IU/mL) and treated with 50 ╬╝g of L-thyroxine. His symptoms improved, and he was discharged 2 weeks after hospitalization. The patient was advised to be readmitted for the evaluation of hypothyroidism and short stature. However, his hypothyroidism was treated at a local medical center irregularly at his own discretion, and he no longer visited our hospital.
On admission at 24 years of age, we measured 6 anterior pituitary hormones as well as the target hormone in the morning with fasting status. A thyroid function test revealed free T4 levels of 0.78 ng/dL (normal, 0.8ŌĆō1.0 ng/dL) and TSH levels of 1.45 ╬╝IU/mL (normal, 0.3ŌĆō4.0 ╬╝IU/mL) upon administration of 50 ╬╝g of L-thyroxine. Laboratory examinations indicated a gonadotropin deficiency, showing a reduction in serum concentration of luteinizing hormone (LH) to 0.21 mIU/mL (normal, 1.5ŌĆō9 mIU/mL) and FSH to 1.1 mIU/mL (normal, 2.0ŌĆō9.2 mIU/mL). Fasting morning serum cortisol level was 5.59 ╬╝g/dL and insulin-like growth factor 1 level was 30.5 ng/mL (normal, 83ŌĆō344 ng/mL), which was also markedly low. These findings may suggest panhypopituitarism. To confirm the patientŌĆÖs disease, a combined pituitary function test and sella MRI were performed.
Combined pituitary function test was done. The patient was administered IV infusion with insulin (regular insulin, 0.1 IU/kg), thyrotropin-releasing hormone (TRH) (Protirelin tartrate 700 ╬╝g, Cerenyl, Bio & Chemical R&D, Seoul, Korea) and gonadotropin-releasing hormone (GnRH) (gonadorelin 100 ╬╝g, Relefact LH-RH, Handok Pharmaceutical, Seoul, Korea) from separate syringes. Blood samples were collected at intervals of 0, 30, 60, 90, and 120 minutes for assay of the respective hormones. Combined pituitary function test showed decreased growth hormone (peak GH, 0.06 ╬╝g/L; normal range, >5 ╬╝g/L) and cortisol (peak cortisol, 10.7 ╬╝g/dL; normal range, >22 ╬╝g/dL) secretion to insulin-induced hypoglycemia. This is compatible with complete growth hormone deficiency (GHD) and adrenocortical insufficiency. TSH (peak TSH, 10.48 uIU/mL; normal range, >10ŌĆō30 ╬╝IU/mL) showed partial response to TRH. Peak LH and FSH were 1.96 mIU/mL and 2.39 mIU/mL, respectively, following stimulation with GnRH. This result indicated that the patient was diagnosed with multiple pituitary hormone deficits (Table 1).
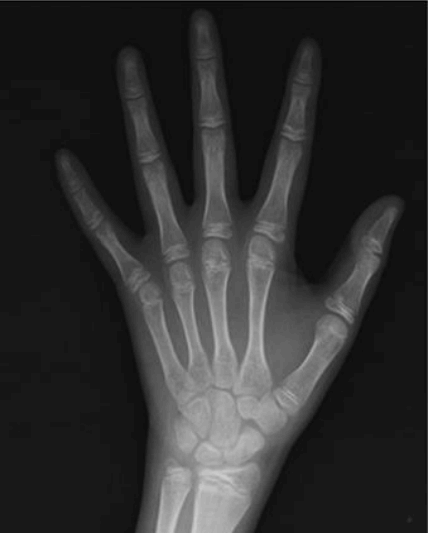
We checked the left-hand anteroposterior view. The patientŌĆÖs bone age, which we determined was between 14 and 15 years using the standards of the Greulich-Pyle method and delayed when compared to his chronological age of 24 years (Fig. 2). His MRI findings showed an absence of pituitary stalk, hypoplasia of the adenohypophysis, and an absence of posterior pituitary gland, indicative of pituitary stalk syndrome (Fig. 3). The patient was treated with hydrocortisone, and his hyponatremia was quickly normalized in response to medication during both the first and second hospitalizations (Fig.4). L-thyroxine was increased from 50 to 75 ╬╝g, and growth hormone therapy was also started.
Written informed consent was obtained from the patient.
Discussion
Hyponatremia is one of the most common electrolyte imbalances; however, symptoms may vary depending on underlying etiology. Some cases can be asymptomatic, but severe hyponatremia can be fatal. Bartter and Schwartz [7] described patients with SIADH who have an ŌĆśinappropriate vasopressin secretion' despite euvolemia. SIADH excludes causes by hypopituitarism and adrenal insufficiency, but there are some cases similar to SIADH in patients with hypopituitarism [8]. Diederich et al. [6] describe the patients as ŌĆśSIADH-like syndrome.ŌĆÖ
Glucocorticoid physiologically suppresses corticotropinreleasing hormones and vasopressin secretion in hypothalamic neurons [9,10]. Furthermore some studies reported that hyponatremia with hypopituitarism developed in patients because of failure in the suppression of vasopressin secretion due to hypocortisolism [11,12]. This hyponatremia occurs commonly in ACTH deficient patients without diabetes insipidus. When glucocorticoid therapy is started, diabetes insipidus may become manifest because of suppressing vasopressin by hydrocortisone and enables normal electrolyte-free water excretion [6,13].
At the age of 14 years old, the patient had drowsy mentality and hypotension which implied adrenal crisis. On the second admission, he did not show any symptoms of adrenal crisis, but he suffered from euvolemic hyponatremia and was also under an acute stress event such as cholecystitis. The patient could not overcome a normal physiological response with increase of endogenous cortisol production in both situations. With the failure to increase endogenous cortisol production, hypocortisolism worsened and hyponatremia occurred, similar to SIADH, due to failure to suppress vasopressin. Although the clinical course is similar to that of SIADH, the treatment is completely different. The main treatment of SIADH is fluid management, however, patients with 'SIADH-like syndrome' respond poorly to fluid management and hyponatremia is corrected by hydrocortisone administration. These patients with hypopituitarism also did not develop hyperkalemia and hyperreninemia because although aldosterone secretion may be slightly decreased in hypopituitarism due to ACTH deficiency, the residual aldosterone, which is regulated by the renin/angiotensin system, is enough to maintain normal plasma volume [5]. Our patient also developed diabetes insipidus when glucocorticoid therapy (at a dose of 10 mg 2 times per day) was started. He was administered DDAVP (1-desamino-8-D-arginine vasopressin) nasally at doses of 10 ╬╝g/0.1 mL 2 times per day and his symptoms improved. Since then, hydrocortisone was continued at a dose of 10 mg 2 times per day. Deodati and Cianfarani [14] have been reported that adult height is considered achieved when a growth rate is <1.5 cm/yr, or bone age >15 years in girls and >16 years in boys. Some studies reported that testosterone might advance bone age [15,16]. At a height of 146 cm, the patient had a very short stature, considering his age. Therefore, growth hormone therapy (at a dose of 0.3 mg/kg/wk) was started at between 14 and 15 years of bone age and testosterone supplementation was held until his bone age reached 16 years old. Recombinant human GH (rhGH) was started with conventional dose (0.3mg/kg/wk) used in patient with GHD [17]. One study reported that high dose rhGH treatment in patient with GHD in puberty increased final height [18]. After several months of rhGH use, if the response is not adequate, we also have plan to increase up to high dose (0.7 mg/kg/wk).
The pathogenesis of PSIS remains elusive. Traumatic birth was suggestive of the etiology, considering the high frequency of perinatal events in patients with PSIS. Recent studies had reported organogenesis defects, especially midline defects, suggesting genetic alterations as the underlying mechanism [19]. Most cases of PSIS are sporadic and occasionally familial, because most patients with PSIS have hypogonadism [20]. Our patient is also considered sporadic because his motherŌĆÖs height was about 155 cm and father's height was 167 cm and they denied hospitalization due to serious illness. Their appearance also looked normal. Mutations of transcriptional factor genes associated with pituitary development have been reported. The genes PROP1, LHX3/LHX4, PIT1, PROKR2, OTX2, HESX1 and TGIF were reported in some studies [19,20]. Unfortunately, we had no genetic testing results.
The true prevalence of hyponatremia associated with cortisol deficiency is unknown. Cuesta et al. [12] reported 573 patients with SIADH. Of the 573 patients, 22 (3.8%) were initially diagnosed with SIADH, then reclassified having secondary adrenal insufficiency based on laboratory findings and clinical presentation. The author also emphasized that adrenal insufficiency might be an underrecognized cause of hyponatremia. Secondary adrenal insufficiency should be highly suspected in patients with hyponatremia, but without hyperkalemia. Verbalis et al. [13] recommends measuring cortisol at 9 AM in hyponatremia. The level of cortisol is under 10 ╬╝g/dL, then is not physiological in acutely ill patients. Secondary adrenal insufficiency should be primarily verified in these patients.
Some cases of hyponatremic hypopituitarism have been reported in Korea, but most of the reports are associated with Sheehan's syndrome, hemorrhagic fever, and brain masses in adults.
To the best of our knowledge, this patient is the first case of PSIS in Korea that occurred during adolescence and presented severe hyponatremia associated with secondary adrenal insufficiency as the first symptom.