Introduction
Noonan syndrome (NS, OMIM 163950) is an autosomal dominant disorder associated with germline mutations in genes coding for components of the RAS/mitogen-activated protein kinase (RAS/MAPK) pathway [1]. NS presents with multiple congenital anomalies and is characterized by short stature, dysmorphic facial features, congenital heart defects, and developmental delay [1,2]. Other clinical findings are a broad and webbed neck, sternal malformation (pectus carinatum or pectus excavatum), scoliosis, gastrointestinal and feeding issues, renal anomalies, cryptorchidism, coagulation defects, and lymphatic dysplasia [3]. The traditional diagnostic criteria of NS are defined as follows: (1) having a typical face with dysmorphic features along with one other major or 2 minor clinical characteristics or (2) having a suggestive face with two major or three minor clinical characteristics [4]. However, some patients have subtle dysmorphic features and may require evaluation by an experienced dysmorphologist [5]. Because NS shows considerable clinical and genetic heterogeneity, both clinical suspicion and genetic testing are important in its diagnosis [1,3].
A pathogenic variant of the genes related to the RAS/MAPK signal transduction pathway is found in approximately 60% of NS cases meeting the traditional clinical criteria [1]. The RAS/MAPK pathway has several genes, the malfunction of which cause RASopathies, including NS and other related disorders with overlapping phenotypic features. Tartaglia et al. [6] first described the association between NS and mutations in PTPN11; it encodes the protein tyrosine phosphatase SHP-2. SHP-2 is a known signaling molecule in the RAS/MAPK pathway. Since then, pathogenic variants of the following genes have been reported to cause NS and other RASopathies: SOS1, RAF1, KRAS, RIT1, NRAS, BRAF, MAP2K1 (MEK1), MAP2K2 (MEK2), SHOC2, CBL, RRAS, RASA2, SOS2, A2ML1, and LZTR1 [1,2,7,8]. Mutation in KRAS is implicated in only approximately 2%ŌĆō3% of NS cases and causes other related disorders, such as cardiofaciocutaneous (CFC) syndrome and Costello syndrome [2,3,9].
Because NS shows genetic heterogeneity, the conventional diagnostic protocol with Sanger sequencing requires a multistep approach [7]. Depending on the frequency of genes associated with NS and their influence on characteristic features, Sanger sequencing of PTPN11 is generally considered the first diagnostic step, followed by that of other related genes of the RAS/MAPK pathway, such as SOS1, RAF1, and KRAS [3,8]. However, if the mutations are in genes other than PTPN11, it becomes more time-consuming and expensive to use the conventional protocol to identify their pathogenic variants in patients. We report a patient who was diagnosed as having NS with a de novo mutation in KRAS identified by targeted exome sequencing (TES). The patient showed several characteristic clinical features of NS, including short stature, dysmorphic facial features, feeding intolerance, cryptorchidism, and intellectual disability.
Case report
A 4-day-old male infant was transferred to the neonatal intensive care unit due to respiratory distress. He was born at 38 gestational weeks to healthy, nonconsanguineous Korean parents. His birth weight was 3,900 g (>90th percentile), and his birth height was 48 cm (25thŌĆō50th percentile). He was intubated and given ventilator support for 3 weeks with empirical antibiotic treatment of ampicillin and cefotaxime; however, no pathogenic organism was detected, and no abnormal findings on chest X-ray were evident. Feeding intolerance was found with poor sucking ability and recurrent vomiting during admission. To determine the possible underlying etiology that resulted in respiratory distress and feeding intolerance, laboratory tests (blood chemistry, thyroid hormone profile, and newborn screening for inborn errors of metabolism using tandem mass spectrometry) and an echocardiogram test were performed; all results were normal.
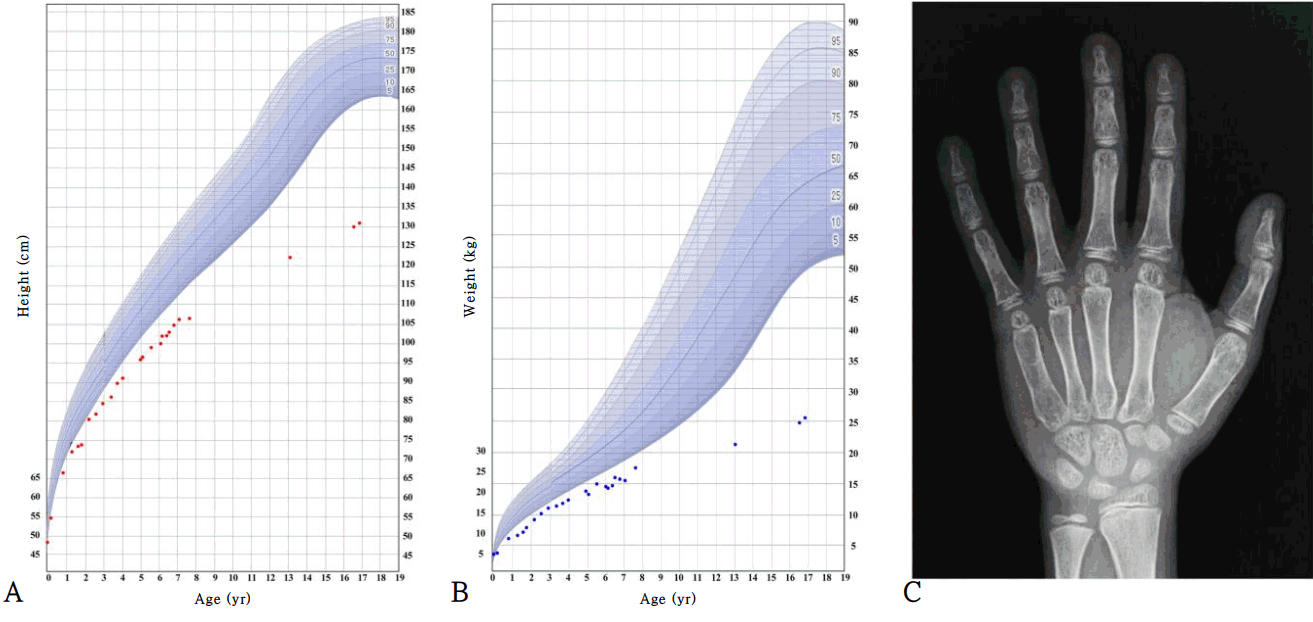
At 15 months of age, he was admitted because of recurrent diarrhea and weight loss. His height was 72 cm (-2.6 standard deviation score [SDS]), and his weight was 6.7 kg (-4.2 SDS) (Fig. 1). Dysmorphic facial features (frontal bossing with a triangular face, mild hypertelorism, depressed nasal bridge, and low-set, posteriorly rotated ears) that were considered suggestive of NS were noted. No palpable mass was found in the abdomen on physical examination. There were no distinctive features of chest wall deformity or other associated anomalies. The karyotype was 46, XY. Abdomen ultrasonography revealed intussusception of the ileocolic type, and a right undescended testicle was incidentally found in the right inguinal sac. Because air reduction had failed to resolve the intussusception, manual reduction of intussusception and orchiopexy of the right testicle were performed. One month postoperatively, his feeding intolerance and diarrhea resolved.
At 6 years of age, his height was 99.8 cm (-3.5 SDS), and weight was 14.4 kg (-3.9 SDS). Growth hormone (GH) stimulation tests were performed, and no GH deficiency was identified. The peak GH levels provoked by the levodopa, glucagon, and insulin tolerance tests were 6.46, 8.98, and 12.40 ng/mL, respectively. Due to concerns regarding his persistent short stature, GH therapy was started at a dose of 0.24 mg/kg/wk. However, the patient gained only 3 cm during 6 months of treatment (Fig. 1). Thus, after 6 months, his parents did not want to continue GH therapy; thereafter, he received no medical assistance for short stature.
At 16 years of age, he visited the hospital again due to short stature and delayed puberty. He showed short stature with Tanner stage II of the genital area (testis 4 mL). His height was 130.1 cm (-5.0 SDS), and weight was 24.8 kg (-4.8 SDS) (Fig. 1). The heights of his father and mother were 170 cm and 155 cm, respectively (the midparental height was 169 cm). Upon laboratory analysis, the levels of insulin-like growth factor 1 and insulin-like growth factor-binding protein 3 were 125.1 ng/mL (-1.8 SDS) and 1,957 ng/mL (-2.0 SDS), respectively. The testosterone level was 0.12 ng/mL (compatible with Tanner stage I), and the peak luteinizing hormone and follicle-stimulating hormone levels from gonadotropin-releasing hormone stimulation were 13.7 ng/mL and 6.21 ng/mL, respectively. The thyroid function test and basal cortisol results were normal. The bone maturation was severely delayed (Fig. 1, his chronological age was 16 years, but his bone age was 10 years for a male according to the Greulich and Pyle atlas), while a skeletal survey showed no remarkable findings. Bone densitometry showed osteoporosis of the lumbar spine (z-score, -4.7, according to gender-specific and age-matched Korean pediatric reference data [10]) and femur (z-score, -3.5). His intelligence quotient was <45 according to the Korean Wechsler Adult Intelligence Scale and 32 according to the Visual-Motor Integration Test.
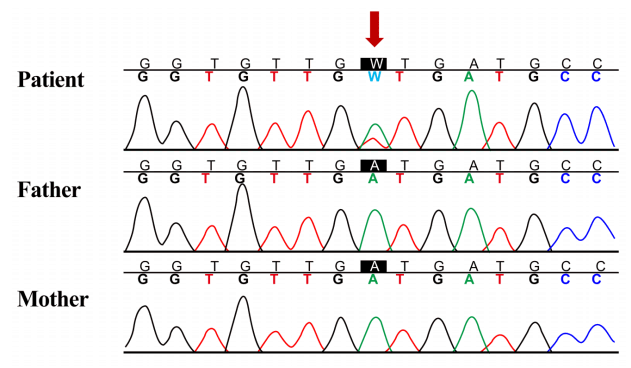
In order to make a diagnosis, TES was performed to find the pathogenic variants of multiple genes associated with the RAS/MAPK pathway. Genomic DNA was extracted from the peripheral blood. Library preparation was conducted by using the TruSight One Sequencing Panel (Illumina Inc., San Diego, CA, USA), which enriches a 12-Mb region spanning 4,813 genes with clinical relevance. Massively parallel sequencing was performed on the Illumina NextSeq platform. The results of exome sequencing revealed a heterozygous de novo mutation of c.458A > T (D153V) in exon 5 of KRAS (reference sequence: NM_004985.4), which was confirmed via direct sequencing (Fig. 2), and there were no pathogenic variants of other genes that cause NS. The D153V variant of KRAS has been reported in patients with NS [2], and parents did not have the same variant.
Finally, the patient was diagnosed as having NS. Careful genetic counseling about NS was provided to the patient and his parents. On ophthalmologic examination, mild strabismus and latent nystagmus were noted. However, visual acuity was normal, and ptosis was not found. He had no hearing problems. GH treatment with an approved dose for NS (0.45 mg/kg/wk) was restarted.
This study was approved by the Institutional Review Board of Samsung Medical Center (approval number: 2012-05-080-008). Informed consent was obtained from the patient and both parents.
Discussion
Patients with NS have clinically and genetically heterogeneous features. Characteristic facial features include a large head with a small face, hypertelorism, ptosis, a depressed nasal bridge, lowset, posteriorly rotated ears, and a deeply grooved philtrum [1]. To fulfill the diagnostic criteria of NS, a typical or suggestive facial appearance with certain characteristic clinical features is needed [4]. Even if the clinical findings meet the diagnostic criteria, a multistep approach is required for the molecular diagnosis of NS and other RASopathies. Moreover, even patients whose clinical findings are insufficient for the diagnosis of NS have pathogenic variants of genes associated with the RAS/MAPK pathway [7]. Many genotype-phenotype correlations in NS and other RASopathies have been made; however, no exclusive phenotype has been found among specific genotypes [1,11].
The conventional strategy for molecular genetic diagnosis of NS is Sanger sequencing of the first candidate gene, which is decided by a close evaluation of the clinical features and consultation with a molecular geneticist, followed by Sanger sequencing of other related genes of the RAS/MAPK pathway in the decreasing order of relevance. Molecular characterization by the conventional method was very expensive and timeconsuming. Lepri et al. [7] conducted TES for 80 patients clinically suspected as having RASopathies and found 38 mutations in 6 of 11 RAS/MAPK pathway genes. A comparison between TES and Sanger sequencing showed the superiority of TES in terms of cost and duration. In this study, TES was performed to screen for the pathogenic variants of multiple genes associated with NS or other related generic disorders resulting in short stature, dysmorphic facial features, delayed puberty, and intellectual disability [12-15]. A heterozygous de novo mutation of c.458A > T (D153V) in exon 5 of KRAS was identified by TES.
Mutations in KRAS have been found in patients with NS, CFC, and Costello syndrome and are associated with severe intellectual disability, longstanding feeding problems, and failure to thrive [2,5,9]. The D153V mutation of KRAS identified in this case has been reported in severe NS or CFC patients who had characteristic facial features, short stature, pectus excavatum, ocular problems, and mild developmental delay [2,9,16]. CFC is known to have similar clinical features to NS, but it is distinguished from NS by ectodermal abnormalities [16]. Cryptorchidism was found in 2 patients. Two of 3 patients had congenital heart diseases [2,9]. The patient in this case had extreme short stature since infancy along with characteristic facial features suggestive of NS, gastrointestinal problems, ocular problems, and intellectual disability. Feeding difficulties and gastroesophageal reflux are common in patients with NS and typically resolve by around 18 months of age [17]. There have been a few reports of gut malrotation [18]; however, intussusception has never been reported in previous studies of NS [1,18]. Even though the patient showed extreme short stature with characteristic facial features suggestive of NS, the diagnosis was delayed due to the absence of congenital heart defects in the patientŌĆÖs infancy, which otherwise typically occur in 50%ŌĆō80% of patients with NS and are a major medical concern for patients and clinicians [1].
Two Korean patients have been reported as having NS with a mutation (V14I and Q23R) in KRAS [3,19]. The former patient was diagnosed in the neonatal period and had pulmonary stenosis, strabismus, and developmental delay [3]. The latter was diagnosed in infancy and had patent ductus arteriosus, an atrial septal defect, hypertrophic cardiomyopathy, ptosis, cystic lymphangioma, and feeding difficulty [19]. In these cases, early diagnosis of NS was possible because they had characteristic clinical features, including cardiac problems. Our patient showed severe short stature with delayed bone maturation and sexual development before the diagnosis of NS was confirmed. Considering the midparental height was 169 cm and there was no parental history of constitutional delayed puberty or growth, he was initially suspected of having an underlying genetic cause for his short stature and delayed puberty and was later diagnosed as having NS. Therefore, we expect some improvement of height with a higher dose of GH (0.45 mg/kg/wk) approved for NS, even at the age of 16, and are considering pubertal induction by testosterone injection through careful discussion with the patient and his parents.
With TES, we found a pathogenic variant of KRAS in a patient with an atypical NS phenotype. In addition to close and accurate evaluation of the clinical features, using TES in patients clinically suspected of having NS facilitates a less expensive and faster molecular diagnosis of NS compared with the conventional Sanger sequencing analysis. Furthermore, since molecular genetic diagnosis with a next-generation sequencing panel that covers multiple genes of the RAS/MAPK pathway will be covered by the National Health Insurance Service in Korea in 2017, the molecular approach to the diagnosis of NS is expected to become easier in the future.