Introduction
Cushing syndrome (CS) is a rare disease in children and adolescents, and the diagnosis and management of pediatric CS are very challenging. CS is a state of chronic hypercortisolism caused by exogenous or endogenous exposure to glucocorticoids [1]. The overall incidence of the disease is about 0.7ã2.4 cases per million persons per year, approximately 10% of which occur in children [2,3]. In infants and children <7 years of age, adrenal causes of CS are the most common, whereas Cushing disease is a common finding in children >7 years of age, accounting for 75%ã80% of all cases of endogenous CS [1,3]. Ectopic adrenocorticotropic hormone (ACTH) production and tumors that produce corticotropin-releasing hormone (CRH) are very rare in the pediatric group [2,3]. Since the most common symptom of pediatric CS is weight gain accompanied by a lack of height gain despite insidious onset, the review of a patient's growth chart is key to distinguishing pediatric CS from simple obesity [1,3,4]. Early diagnosis and treatment of CS can lead to a decrease in disease-associated morbidity during childhood [4]. After laboratory confirmation of hypercortisolism, localization, and lateralization are necessary for the diagnosis of CS. A pituitary magnetic resonance imaging (MRI) is a useful diagnostic imaging modality; in addition, bilateral inferior petrosal sinus sampling (IPSS) after CRH administration is helpful in identifying ACTH-secreting adenomas, which are invisible on MRI scans [3-6]. The treatment of choice for almost all patients with Cushing disease is surgical removal through transsphenoidal surgery (TSS). Although the success rate of the first TSS is >90% and the recurrence rate is reported to be about 15% with a range of 11.5%ã47.4% [1,6,7], all patients require ongoing surveillance for consideration of additional treatment after TSS.
In this report, we present the case of a 17-year-old boy with Cushing disease who presented with obesity and vertebral compression fractures, and show multiple pituitary adenoma (MPA) through a second TSS.
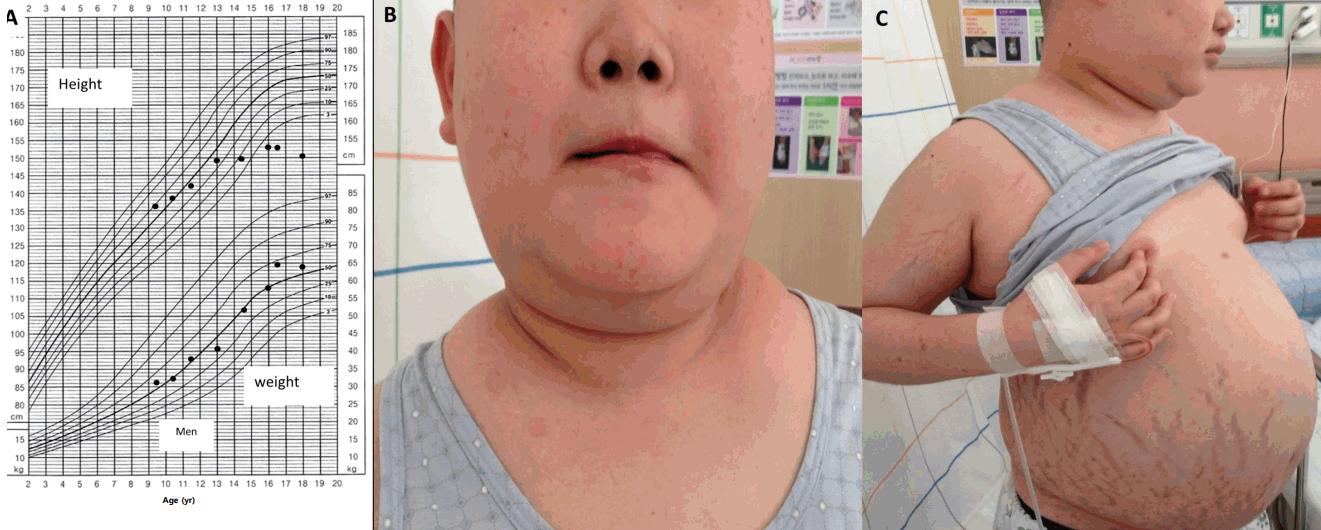
Case report
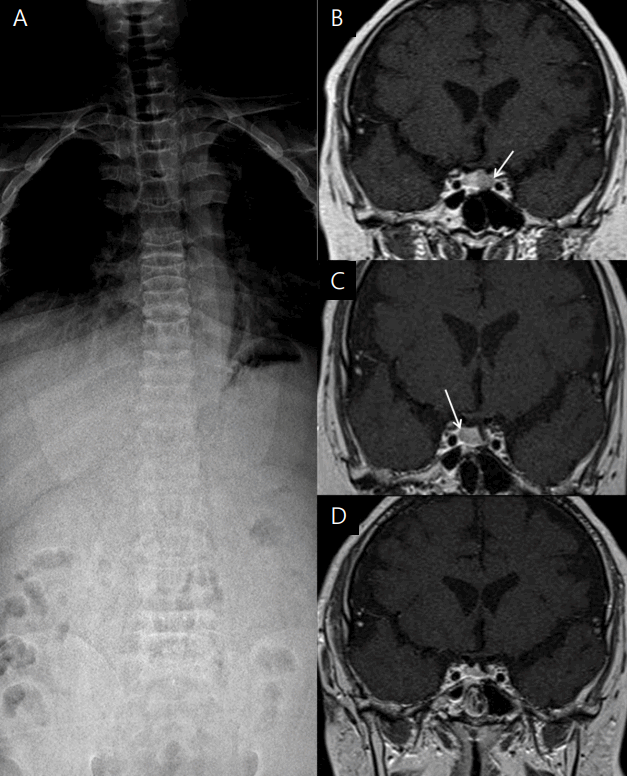
A 17-year-old boy was referred to Pusan National University Children's Hospital for evaluation of a vertebral compression fracture and obesity. The subjectãs height and weight were 149.5 cm (-4.1 standard deviation score [SDS]) and 63.6 kg (-0.1 SDS), respectively. The midparental height was 178 cm (father was 178 cm and mother was 165 cm), and there was no family history of pituitary tumors or developmental delay. His body mass index (BMI) was 28.5 kg/m2 (3.0 SDS). The subject had gained 23 kg, but he had not grown in height over the course of 4 years (Fig. 1A). Since the patient attended a special school owing to an intellectual disability of unknown origin (intelligence quotient, 65), growth failure and obesity were neglected despite typical Cushingoid features, including a buffalo hump, central obese, moon-shaped face, multiple abdominal striae, relatively thin limbs, hypertrichosis, and acanthosis nigricans that had been apparent for 4 years (Fig. 1B, C). A selective serotonin receptor inhibitor had been prescribed for a mood disorder. The testicular volume was 8 mL, and the Tanner stage III pubic hair was observed. The bone age was 2 years behind his chronological age; in addition, a simple X-ray of the whole spine showed osteopenia and multiple compression fractures on the T9, L2, and L3 vertebral bodies (Fig. 2A). Furthermore, T-scores of the bone mineral densiometry (BMD) at lumbar spine and hip were -4.9 and -2.7, respectively.
Plasma ACTH at 8 AM and 24-hour urinary free cortisol (UFC) levels were elevated to 68.5 pg/mL and 228.5 ö¥g/day (normal, <50 ö¥g/day), respectively. Serum cortisol levels at 8 AM and 8 PM were 31.1 ö¥g/dL and 15.1 ö¥g/dL, respectively, which represented a loss of diurnal rhythm (Table 1). The serum cortisol, plasma ACTH, and 24-hour UFC levels after a low-dose dexamethasone suppression test for 2 days (0.5 mg every 6 hours) were 22.9 ö¥g/dL, 86.7 pg/mL, and 1,363.7 ö¥g/day, respectively, and they were not suppressed. A high-dose dexamethasone suppression test for 2 days (2 mg every 6 hours) revealed that the serum cortisol, plasma ACTH, and 24-hour UFC levels were suppressed to 0.75 ö¥g/dL, 27.3 pg/mL, and 5.1 ö¥g/day, respectively. A pituitary MRI suggested an 8-mm microadenoma on the left side of the pituitary gland (Fig. 2A). Bilateral IPSS was performed to confirm the laterality and localization of ACTH secretion from the right or left pituitary gland. The ratio of inferior petrosal sinus (IPS) to the peripheral blood (P) level of ACTH before CRH injection was 9.75 (reference level >2), and the IPS/P after CRH injection was more than 8.67 (reference level >3), which indicated the presence of an ACTH-secreting adenoma. However, all ACTH peaks in the patient's left and right sides were more than 1,500 pg/mL and we could not determine the laterality of ACTH secretion (Table 2). During the operation, the surgeon decided to perform a left microadenomectomy; this was because there was no visible tumor in the right pituitary gland and the pathology results of the biopsied tissue were consistent with an ACTH-secreting adenoma. The gross finding of the specimen revealed a yellowwhite soft tissue. Serum cortisol and 24-hour UFC were obtained 2 days after TSS. Morning cortisol and ACTH levels were decreased from 31.1 to 5.7 ö¥g/dL and from 68.5 to 6.7 pg/mL, respectively. However, the 24-hour UFC level was elevated from 228.5 to 302.1 ö¥g/day. After the operation, the persistent headache was improved, and hypertensive drugs could be tapered for a while. However, there was no weight reduction. Three months after TSS, the subject's BMI had not improved, and headache and hypertension were observed again.
Serum cortisol, plasma ACTH, and 24-hour UFC levels were increased to 27.2 ö¥g/dL, 25.4 pg/mL, and 232.2 ö¥g/day, respectively, for 6 months after the left microadenomectomy. A follow-up pituitary MRI revealed a 9-mm microadenoma, which had not been identified in the previous MRI (Fig. 2B), in the right pituitary. The right microadenomectomy was treated with TSS, and the histological findings confirmed an MPA positive for ACTH, growth hormone (GH), thyroid stimulating hormone, gonadotropins (luteinizing hormone and folliclestimulating hormone), and prolactin. The gross finding of the surgical specimen was not different compared to the previous adenoma.
Two days after the second operation, serum cortisol and plasma ACTH levels were consistently elevated to 18.4 ö¥g/dL and 26.6 pg/mL, respectively. Although a follow-up pituitary MRI after the right microadenomectomy demonstrated no evidence of a pituitary tumor (Fig. 2C), serum cortisol, plasma ACTH, and 24-hour UFC levels remained elevated to 17.9 ö¥g/dL, 15.1 pg/mL, and 416.0 ö¥g/day, respectively. A combined pituitary hormone function test was performed twice after each surgery, only revealing a deficiency in GH (Table 3). However, GH therapy was withheld due to the possibility of invisible tumor growth.
Since hypercortisolemia in this subject was not resolved despite a repeated TSS and since no visible tumor in MRI was identified, we recommended that the patientãs parents consider additional options, such as total removal of the pituitary gland, radiotherapy, or bilateral adrenalectomy. However, the patient and his family chose to have a follow-up MRI before deciding on an additional therapeutic option. The patient continued medications of oral calcium carbonate combined with calciferol daily and intravenous bisphosphonate (1 mg/kg/day over 3 consecutive days) every 4 months to treat osteoporosis. There was no additional fracture at the onset of osteoporosis treatment, and the T-scores of BMD at lumbar spine and hip had improved to -4.3 and -2.6, respectively, after 1 year of treatment. The subject was also treated with 3-hydroxy-3-methylglutarylcoenzyme A reductase inhibitor and angiotensin-converting enzyme inhibitor for persistent dyslipidemia and hypertension.
We obtained the written informed consent for publication from the patient and his parents.
Discussion
This report describes an adolescent patient with latediagnosed Cushing disease who showed a compression fracture, rapid weight gain, growth failure, and delay in pubertal progression. This report also shows that the patient needed TSS twice due to 2 distinct pituitary adenomas.
Patients with late-diagnosed CS have more severe medical problems than those who are diagnosed early; these medical problems include cardiovascular risk factors such as diabetes mellitus, hypertension, hyperglycemia, hyperlipidemia, obesity, osteoporosis, and even death [1]. Inadequately treated Cushing disease patients have a 4ã5 times higher mortality rate than the general population [1,8]. Therefore, early diagnosis and treatment of Cushing disease is important for a successful prognosis without complications [1].
In general, a pituitary MRI is the main tool used to confirm the presence or absence of a pituitary adenoma; it should be performed in thin sections with a high resolution and contrast (gadolinium). However, microadenomas are more common in Cushing disease, and a ~50% detection rate in MRIs means it is difficult to identify a definite pituitary tumor [1,3,6]. To obtain an accurate diagnosis, the most direct method is to measure ACTH in the pituitary venous drainage by IPSS [9,10]. IPSS is a highly invasive procedure requiring technical expertise; it has successful catheterization rates that are described in the literature as ranging from 65%ã100% [11]. Since an MRI failed to identify Cushing disease in some patients, bilateral IPSS could be helpful for lateralization of a tumor together with close monitoring of hormone status [1,3,4,9]. According to a recent consensus, IPSS should be performed for a microadenoma less than 6 mm in a pituitary MRI [9]. IPSS can also detect an occult ectopic ACTH secreting tumor [9,10]. In our subject, bilateral IPSS markedly showed ACTH secretion from the right and left sideãdespite the visibility of only an 8-mm-sized left pituitary microadenoma on a brain MRI scan. Furthermore, a newly developed right pituitary microadenoma was visible on a repeat brain MRI taken 6 months after the first surgery. The latter microadenoma was confirmed with MPA; however, only the clinical manifestation of hypercortisolemia was present in our case. MPA are defined as two more immunocytochemically and/or morphologically distinct tumors that are detected in only 0.37%ã2.6% of surgical specimens and in 1.6%ã3.3% of Cushing disease patients [12,13]. Therefore, pathologic examination after surgical removal is important for definite diagnosis of MPA [12]. MPA are frequently nonfunctional and small-sized, and they show low imaging detection and high surgical failure [12,13]. However, the most common clinical presentation is CS (38%) and then acromegaly (34.9%) for 64 patients with MPAs [12]. Since limited information is available for genetic or epigenetic changes in MPA, the pathogenesis of MPA remains elusive [12].
Selective adenomectomy by TSS is the treatment of choice for Cushingãs disease [1-4,8]. The remission and recurrence rates after TSS have been reported to be in the ranges of 65%ã95% and 8%ã35%, respectively [14-16]. Several factors may influence remission in Cushing disease, such as preoperative ACTH level, UFC levels, adenoma size, localization on MRI, intraoperative tumor visualization, dural invasion, histological confirmation of corticotroph adenomas, skillful surgery, and age of patient. Monitoring levels of early morning serum cortisol and 24-hour UFC are most commonly used when determining remission or recurrence after TSS [7]. A biochemical cure was defined using a morning serum cortisol level; a cutoff of 5 ö¥g/dL was most widely employed despite a range from 5ã10 ö¥g/dL (50ã275.9 nmol/L) during the first week after surgery [6-8,14,16]. Since the serum nadir cortisol after initial TSS slightly exceeded 5 ö¥g/dL and since IPSS before surgery revealed no laterality in this subject, it is difficult to define "incomplete resection" or "recurrence" in our case. Because we also administered a stress dose of hydrocortisone immediately after surgery, free UFC is not a reliable marker to assess endogenous cortisol status. On radiologic findings, recurrence after the initial TSS was apparent; unfortunately, the second operation showed intermediate failure to reduce hypercortisolism despite inconclusive MRI findings. Since IPSS results were not compatible with imaging at diagnosis and the interval was too short to identify a new adenoma after surgery, occult MPA might have been present at the moment of diagnosis of Cushing disease. Periodic imaging follow-up will be needed in this subject.
In addition to the possibility of incomplete resection of an invisible pituitary adenoma, tumor invasion in the cavernous sinus could be present in this subject. A recent report showed ectopic ACTH-secreting pituitary adenomas, which were found in patients with bilateral peak ACTH on IPSS. The report also showed invisible ACTH-secreting adenomas in patients who were diagnosed with ectopic ACTH-secreting pituitary adenomas in the cavernous sinus or ethmoid sinus [17]. In our case, a review of the MRI scans showed no identifiable mass on the follow-up pituitary MRI, and additional treatment was needed for persistent hypercortisolemia. Persistent or recurrent Cushing disease patients can be managed with alternative treatments such as medical therapy, radiotherapy, or total removal of the adrenal or pituitary glands [2]. As the patient and his family members refused additional brain surgery, we considered medical adrenalectomy, surgical bilateral adrenalectomy, or radiotherapy as treatment options. Bilateral adrenalectomy leads to an immediate reduction in hypercortisolemia, and it requires administration of glucocorticoid and mineralcorticoid hormones as well as monitoring for the possibility of corticotroph tumor progression [15,18]. The success rate of radiotherapy is 40%ã70% without an immediate therapeutic effect; pituitary insufficiency and infertility are generally expected side effects [7,8,15].
In conclusion, we reported the clinical course of Cushing disease with 2 distinct pituitary adenomas. Since there is no consensus as to the best treatment for relapsing or persistent Cushing disease and since only a few cases of MPA among pediatric Cushing disease have been reported, a close followup of tumor status, severity of hypercortisolism, and patientsã perspectives are the major parameters used to determine the best treatment option for each patient. In addition, early recognition and diagnosis of pediatric Cushing disease would lead to earlier recovery, improved growth, and better quality of life.