Introduction
The chromosome 22q11.2 deletion syndrome (22q11DS) is the most common microdeletion syndrome with an estimated incidence of 1 in 4,000 live births1). Heterozygous deletion in chromosome 22q11.2 is found in most patients, and about 10% of patients have microdeletion that was inherited from affected parents1). The main clinical features of 22q11DS include congenital heart defect, hypocalcemia due to hypoparathyroidism, thymic hypoplasia, palatal cleft and mental retardation1,2). Hypoparathyroidism is most likely detected from symptoms of hypocalcemia, such as seizure, tremors or tetany that usually manifests during the neonatal period, and neonatal hypocalcemia is usually considered as one of the first manifestation of 22q11DS2). The hypocalcemia can be transient, although it may recur later in life. Late onset hypocalcemia in adolescence or adulthood have been reported in patients with 22q11DS due to its highly variable phenotype2,3).
The extreme diversity of clinical presentations makes 22q11DS without well-known phenotype a diagnostic challenge. Because of possible underdiagnoses, true prevalence of this syndrome can be much higher than reported4,5). We hereby report the case of a girl with the history of imperforate anus, partial cleft palate, and mild mental retardation, but without history of neonatal hypocalcemia or major cardiac anomaly, who was diagnosed for 22q11DS at the age of 11 after the onset of overt hypocalcemia.
Case report
An 11-year-old girl visited our Emergency Department with fever, cough, abdominal pain and vomiting. Hypocalcemia (serum calcium, 5.0 mg/dL; ionized calcium, 0.74 mM) was noted on laboratory analysis, with normal albumin (4.3 mg/dL), and upper normal limit level of phosphorus (5.6 mg/dL).
She was born with 3,040 g of body weight at 37 weeks of pregnancy at our hospital and was the second child of phenotypically normal Korean parents. Imperforate anus with rectovestibular fistula and partial cleft palate were found at birth. Atrial septal defect (ASD, 3.5-mm width) was diagnosed on echocardiography performed on the first day of life. However, the karyotyping revealed normal chromosomal pattern by GTG banding. Descending colostomy was performed on the first day of life and followed by colostomy repair at 9 months of age. Pena operation (posterior sagittal anorectoplasty) was performed at 4 months of age and transposition anoplasty was done at 7 months of age. Palatoplasty for cleft palate was performed at 14 months of age, and again at 9 years of age, velopharyngeal insufficiency was surgically corrected by superiorly based pharyngeal flap with lateral port control. Echocardiography at 20 months of age showed no intracardiac anomaly, suggesting spontaneous closure of previously observed ASD. She had suffered from frequent respiratory tract infections with otitis media and chronic constipation, but there was no history of severe systemic infection. Her developmental milestones were delayed, and she was diagnosed for mild intellectual disability at 8 years of age (Intelligence quotient 57 on Korean Wechsler intelligence scale for children). She was attending a public school without specific behavioral problems, although her academic performance was poor.
On the third day of life, her calcium (9.2 mg/dL), phosphorus (4.5 mg/dL), and ionized calcium (1.05 mM/L) levels were normal. Her calcium level was also normal on preoperative screening before surgery at 4, 7, and 9 months of age (10.5, 10.8, and 10.6 mg/dL, respectively). Her calcium level was in low normal range on laboratory studies at 20 months and 7 years of age (9.1 and 8.9 mg/dL, respectively). The parents denied any history of hypocalcemic symptoms during her infancy or childhood.
Her facial features appeared mildly dysmorphic, with hypertelorism, short philtrum and small down-turned mouth. Hypernasal speech was not observed. At the time of hypocalcemia onset at 11 years of age, her height was 141 cm (25th percentile) with weight 31 kg (10thŌĆō25th percentile). Her midparental height was 157 cm (10thŌĆō25th percentile). Her wrist X-ray showed bone age of 12 years without any evidence of rickets. Serum magnesium level was normal (1.63 mg/dL). The parathyroid hormone (PTH) level was inappropriately low (10.8 pg/mL; reference rage, 15ŌĆō65 pg/mL) considering her plasma calcium level, suggesting hypocalcemia due to hypoparathyroidism. Serum 25(OH)D level was also decreased (11.4 ng/mL; reference range, 30ŌĆō100 mg/mL). Her thyroid function was normal (thyroid-stimulating hormone, 0.28 uIU/mL; free T4, 1.68 pg/mL).
Intravenous calcium (calcium gluconate, 100 mg/kg) was given during the first 3 days after admission, followed by oral calcium (calcium lactate, 300 mg/kg/day). Vitamin D (calcitriol, 0.75 ┬Ąg/day) treatment was started on the 2nd hospital day. She was discharged on the 6th hospital day when her calcium level was 6.5 mg/dL. Her calcium level increased to 8.3 mg/dL on follow-up visit at 1 week after discharge, and her PTH level was still low (9.0 pg/mL). Although 25(OH)D level has been normalized (25.5 and 30.5 ng/mL after 2 months and 3 months, respectively) after treatment, daily administration of calcium (calcium carbonate, 62.5 mg/kg/day) and calcitriol (0.5 ┬Ąg/day) was required to maintain normocalcemia (serum calcium 9.2 and 8.3 mg/dL after 2 and 3 months, respectively).
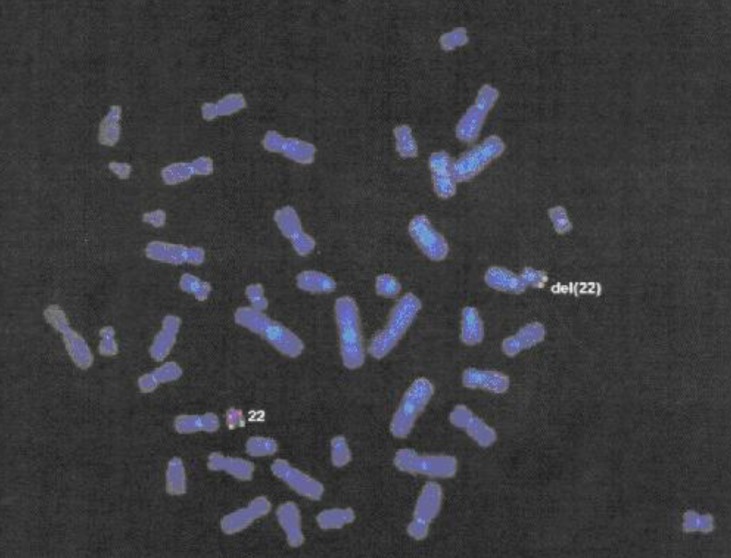
22q11DS was suspected based on her history of velopharyngeal insufficiency and mental retardation, and fluorescence in situ hybridization analysis confirmed a deletion of chromosome 22q11.2 (Fig. 1).
Discussion
The present case suggests that the diagnosis of 22q11DS can be delayed in those without major clinical features. Our patient did not have neonatal hypocalcemia or major cardiac anomaly, although she had imperforate anus, velopharyngeal insufficiency, and delayed development, and subtle but characteristic facial features of 22q11DS5).
The chromosome 22q11 region is very unstable, and misalignment of chromosome-specific low-copy repeats (LCR22A-H) during nonallelic homologous recombination can lead to the deletion of the 22q11.2 region6). Approximately 90% of patients with 22q11DS have the 3Mb (LCR22A-D) deletion, while less than 10% have shorter (~1.5Mb) deletion such as LCR22A-B deletion 6). The haploinsufficiency of genes located at 22q11.2, especially TBX1, can disturb the early morphogenesis of many organs including parathyroid gland, thymus and facial structures6). The 22q11DS encompasses very wide clinical spectrum, including DiGeorge syndrome, velocardiofacial syndrome, and conotruncal anomaly face syndrome5).
Neonatal hypocalcemia due to hypoparathyroidism has traditionally been known as the first manifestation that has been reported in 43%ŌĆō60% of patients with 22q11DS2,7). However, in our case, the patient had obviously normal calcium levels throughout her infancy, and overt hypocalcemia was first noticed at 11 years of age. Low normal range of calcium levels during her early childhood could be a sign of subclinical hypoparathyroidism, but PTH level was not checked before the onset of overt hypocalcemia.
A recent study including 138 adults with 22q11DS reported 80% of patients have a lifetime history of hypocalcaemia8). The spectrum of parathyroid gland function in 22q11DS ranges from severe neonatal hypocalcemia to latent hypoparathyroidism2,8). The PTH secretory reserve is inadequate compared with normal population in patients with latent hypoparathyroidism3), and it becomes insufficient that can lead to evident hypocalcemia when calcium requirement increases such as during adolescence, pregnancy, surgery, infection, or any physiologic stresses2,9).
Hypocalcemia is one of the unique features of proximal deletions (LCR22A-D or LCR22A-B) in patients with 22q11DS6). However, it manifests with a wide clinical spectrum even in the same family ranging from hypocalcemic hypoparathyroidism to normocalcemia with latent hypoparathyroidism, although the latent hypoparathyroidism can evolve to frank hypocalcemic hypoparathyroidism in adulthood10). A transition from subclinical to overt hypoparathyroidism in a child with 22q11DS has also been reported11).
A provocation test using sodium bicarbonate infusion was reported to be able to evaluate residual parathyroid function in normocalcemic patients with 22q11DS8). Hypocalcemia may also be worsened by carbonated beverages such as colas9). Regular lifelong follow-up of calcium, magnesium, and PTH levels are required in patients with 22q11DS12). Calcium and vitamin D supplements are recommended to patients with 22q11DS, regardless of whether they had been diagnosed with hypocalcaemia, however, iatrogenic hypercalcemia, which can result in renal calculi and renal failure, should be avoided9).
Vitamin D deficiency is very common in Korean children, but calcium and PTH levels were normal in most children with vitamin D deficiency13). Our patient also had vitamin D deficiency at the time of hypocalcemia onset. It is possible that vitamin D deficiency may have contributed to the development of hypocalcemia in the patient with latent hypoparathyroidism. However, calcium supplement was persistently required after vitamin D replenishment in our patient, suggesting that vitamin D deficiency was not the main cause of hypocalcemia.
Congenital heart defects are one of main clinical feature of 22q11DS. Although serious cardiac anomalies were present in most patients in earlier studies, the prevalence seems to be about 40% according to recent papers9). Conotruncal defects, especially tetralogy of Fallot are the main forms5). Our patient had a small ASD, which was closed spontaneously. The prevalence of ASD was reported as 12% in patients with 22q11DS4).
Table 1 summarizes the incidences of major and minor clinical features of 22q11DS in recent publications 2,4,5,14,15,16,17). The prevalence of each clinical feature is different according to the age of patients and data sources. It seems that the prevalence of hypocalcemia and psychiatric disorders increase with age, whereas the prevalence of cardiac anomalies seems to be higher in the younger age groups. Intellectual disability and dysmorphic face were observed in most patients.
A delay in the diagnosis of 22q11DS with noncardiac symptoms has been reported in a recent study including 228 patients with 22q11DS4). Among them, 71% of patients were diagnosed before 2 years of age, mainly related to the presence of cardiac anomalies and neonatal hypocalcemia. In patients diagnosed after 2 years of age, developmental delay, minor cardiac defects, recurrent infections and facial features led to the diagnosis of 22q11DS4). According to a recent study, 31% of patients with 22q11DS were diagnosed after age 10 years16). The basis for clinical suspicion was diverse, but once brought to attention, additional symptoms such as characteristic facial features and developmental delay were easily noted in most patients16). Although our patient presented some of the characteristic facial features such as hypertelorism, short philtrum, and small down-turned mouth, it was subtle and did not lead to suspicion of 22q11DS before the onset of overt hypocalcemia.
The incidence of anorectal malformation is approximately 1 in 5,000 live births18). More than half of anorectal malformation is associated with other congenital anomalies such as urogenital and cardiovascular anomalies. A number of congenital syndromes, such as Down syndrome, are accompanied with anorectal malformations, however, the 22q11DS is not considered as the major etiologic condition of anorectal malformations18). However, anorectal malformation, including imperforate anus and symptomatic anal stenosis, was reported in 5% of patients with 22q11DS4), and there are some case reports on symptomatic anal anomalies in 22q11DS19). Our patient had undergone multiple major surgeries for the correction of imperforate anus, but 22q11DS was not suspected at that time.
There are several reports on the delayed diagnosis of 22q11DS, and recent cases of 22q11DS diagnosed after 10 years of age are summarized in Table 23,20,21,22,23,24,25,26,27). Most of them (8 of 12) visited the hospital because of hypocalcemic seizure. However, only 16.7% (2 of 12) had documented history of neonatal hypocalcemic events and 50% (6 of 12) had no cardiac abnormalities, suggesting that the diagnosis may be delayed if the patient has neither hypocalcemic symptoms nor cardiac anomalies. Dysmorphic facial features were usually recognized after the diagnosis of 22q11DS, and learning disability and/or mental retardation was present in most patients. In our case, diagnosis was delayed until the onset of overt hypocalcemia, probably due to the absence of major cardiac anomaly and neonatal hypocalcemia.
A correct diagnosis is important in patients with 22q11DS because of the increased risk for later-onset medical and neuropsychiatric problems, including schizophrenia (>20-fold increase), anxiety disorder, epilepsy, and Parkinson disorder9). Many treatable conditions may be anticipated and features may accumulate over time17). The risk of premature mortality, especially sudden and unexpected death, also increases28).
Our case suggest that imperforate anus, without major cardiac anomaly, can be a clinical presentation of 22q11DS, and that 22q11DS should be considered in the differential diagnosis of hypocalcemia in any age because of its wide clinical spectrum. Furthermore, patients with 22q11DS should be informed of the symptoms of hypocalcemia that may develop later, and periodic screening for serum calcium level should be considered.