Introduction
Deletions of the distal 2q37 region involve the last cytogenetic band on the long arm of chromosome 2,1) and the clinical features of terminal microdeletions of 2q37 were first described in 1989 by Gorski et al.2). The incidence is unclear, but more than 115 patients carrying isolated, primarily terminal deletions with breakpoints at or within chromosome 2q37 have been reported1). This disease shows facial dysmorphism, including a prominent forehead; sparse, arched eyebrows; deep-set eyes; midface hypoplasia; a depressed nasal bridge; a thin upper lip; and various pinna anomalies, ranging from fleshy and/or anteverted lobules to microtia3). In addition, brachymetacarpia and brachymetaphalangia, typical signs of Albright hereditary osteodystrophy (AHO), are well described in 2q37 microdeletion syndrome4). In contrast to pseudohypoparathyroidism Ia or pseudopseudohypoparathyroidism, patients with 2q37 deletion syndrome do not have any mutations in GNAS, and they lack renal parathyroid hormone resistance and soft-tissue ossification5).
About 30% of patients with 2q37 deletions show major malformations, including congenital heart disease, gastrointestinal or genitourinary anomalies, and central nervous system malformations3). Affected individuals often have mild to moderate developmental delay, mental retardation, and hypotonia6). Autistic features also occur in association with terminal deletions with breakpoints at any subband of 2q37.7)
Here, we report a patient with 2q37 deletion confirmed by high-resolution cytogenetic analysis (i.e., microarray-based comparative genomic hybridization [array-CGH]). The patient presented with dilated cardiomyopathy (DCMP), short stature, and AHO-like phenotypes as well as characteristic facial features. DCMP associated with 2q37 deletion syndrome has not been reported in the literature.
Case report
The patient was the first child of healthy nonconsanguineous Korean parents. She was born by cesarean section at 35 weeks of gestation with a birth weight of 2,400 g (25th–50th percentile). She showed left microtia and cupping of the right pinna. Conventional chromosome analysis revealed 46, XX. Her younger sister was healthy, and her family history was unremarkable. Showing an asymmetric facial expression, she was diagnosed with right peripheral-type facial nerve palsy at the age of 2 years and the symptoms persisted. She had mild developmental delay. She walked with support at 15 months of age and started speaking single words at 12 months of age. The patient now attends normal school, although she has moderate learning difficulties.
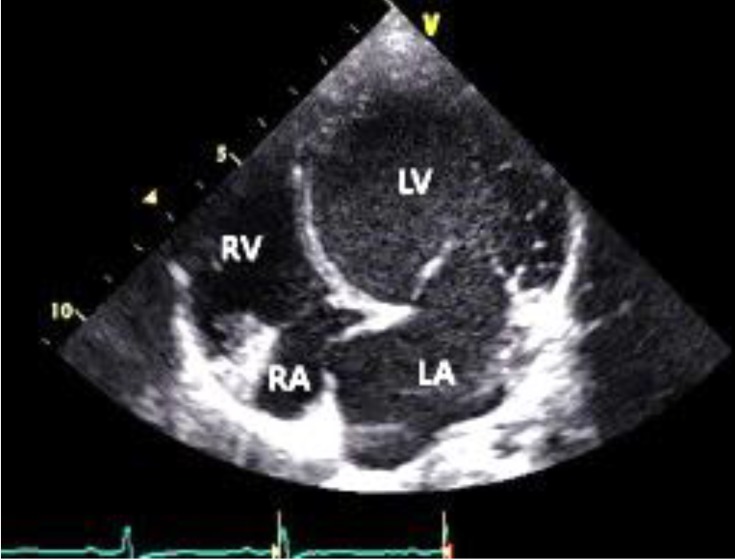
At the age of 7 years, she was admitted to the hospital due to coughing and vomiting. The chest radiograph showed cardiomegaly, and the echocardiogram revealed myocardial dysfunction with a dilated left ventricle. She was diagnosed as having myocarditis with DCMP and heart failure. After three weeks, she recovered with medical treatment. Four months later, she was admitted to the hospital again due to fever, dizziness, and chest discomfort. She was referred to Samsung Medical Center because of her DCMP with congestive heart failure. The echocardiogram revealed severe myocardial dysfunction with a dilated left ventricle (Fig. 1). The estimated left ventricular ejection fraction (EF) was 26%, and the left ventricular end-diastolic dimension was 5.9 cm. Heart magnetic resonance imaging (MRI) revealed DCMP without myocardial fibrosis, and there was no evidence of congenital heart defect or coronary artery anomaly. In addition, a heart biopsy revealed nonspecific findings. The exact cause of the patient's DCMP was unknown. On the 10th day, a follow-up echocardiogram showed improved cardiac function and decreased heart size, and she was discharged with medications (captopril, furosemide, spironolactone, carvedilol, and digoxin).
At the age of 10 years, she visited the outpatient clinic of pediatric endocrinology due to short stature. Given her height of 124.3 cm (-2.25 standard deviation [SD]) and her weight of 24.5 kg (-1.88 SD), her body mass index was 15.9 kg/m2 (-0.26 SD). Her midparental height was 163.5 cm. A physical examination revealed that she was in Tanner stage I for breast development. She exhibited a flat nasal bridge, deep-set eyes, arched eyebrows, and a thin upper lip as well as abnormalities of both ears. Temporal bone computed tomography showed bony atresia of the left external auditory canal, while her hearing evaluation result was normal. In addition, she presented bilateral brachymetatarsia of the third to fifth toes and brachymetacarpia of the third to fifth metacarpal bones (Fig. 2). Ophthalmologic examinations showed congenital myogenic ptosis, and she underwent bilateral levator resection surgery at the age of 10 years. She often showed waddling gait and hyperflexibility of the hips. The patient did not have autistic features.
The data from biological tests (i.e., routine blood cell count, calcium, phosphate, creatinine, serum alkaline phosphatase, 25-hydroxyvitamin D, thyroid hormone, parathyroid hormone, and urine analysis) appeared within the normal ranges. The serum insulin-like growth factor I (IGF-I) value was lower than the age-specific reference ranges (64.9 ng/mL; z-score, -2.4). Radiographs of her hands showed a bone age of 10 years at a chronological age of 10 years and 3 months. The renal ultrasonography results were normal.
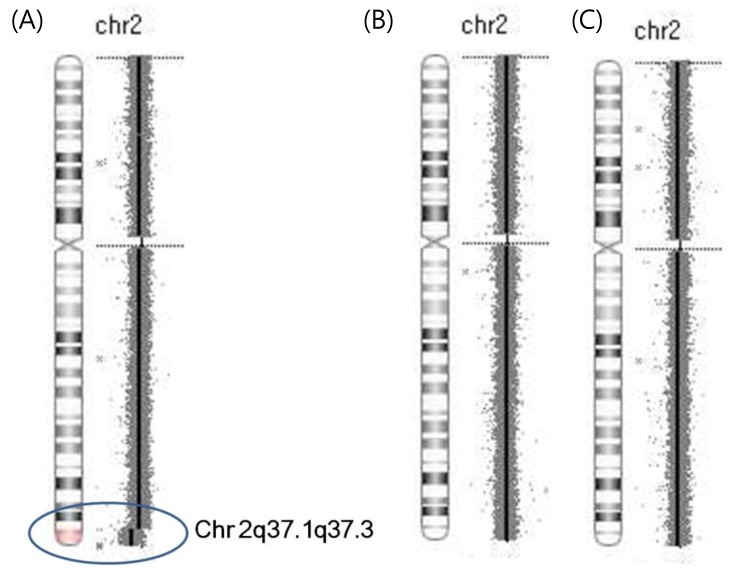
Cytogenetic analysis was performed again using peripheral blood lymphocytes with the 550-band level, including GTG banding, to evaluate her short stature and dysmorphic features. Genomic DNA was isolated from the peripheral blood leukocytes using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). An Affymetrix Cytogenetics Whole-Genome 2.7M Array (Affymetrix Inc., Santa Clara, CA, USA) was used for copy number variation (CNV) analysis according to the manufacturer's instructions. Copy number data were analyzed using the Chromosome Analysis Suite 1.1 (Affymetrix Inc.). The array contains approximately 400,000 single nucleotide polymorphism and 2,370,000 unique nonpolymorphic probes for determination of the CNV. Segments with >50 markers and >100 kb were considered to be CNVs. The hg19 assembly was used as a reference. It revealed the deletion of 2q37.1 to 2qter. Finally, array-CGH revealed terminal deletion (about 8.3 Mb) at 2q37.1q37.3 (Fig. 3). The array-CGH results of both parents were normal.
In the growth hormone (GH) stimulation test, the peak level of GH was 7.56 ng/ml by L-dopa and 8.63 ng/mL by glucagon. Sella MRI revealed a pars intermedia cyst. Human GH therapy (0.55 mg/kg/wk) was started when she was 10 years old, and her growth velocity has improved (Fig. 4). After eight months of GH therapy, her height was 133.1 cm (-0.9 SD) with a significant increase in growth rate to 8.8 cm. In addition, IGF-1 value increased to 266.6 ng/mL after eight months of GH therapy.
The patient did not have any heart failure symptoms while on medications, and her latest echocardiogram at the age of 10 years showed improved cardiac function (an EF of 53.6% and left ventricular end-diastolic dimension of 46.8 mm).
Discussion
We report a Korean patient with deletion of 2q37. In the case of 2q subtelomeric deletions, significant variability in clinical presentation is apparent, but almost all patients have some degree of mental retardation and facial dysmorphism6). Our patient had the following features similar to the reported phenotype of 2q37 deletion: characteristic facial features, ear abnormalities, ptosis, brachydactyly, mild mental retardation, developmental delay, and short stature. Another congenital defect found in our patient was peripheral nerve palsy, but it is nonspecific for the 2q37 deletion phenotype.
Unusually, our patient suffered from DCMP without any congenital structural defects. Congenital heart malformations have been noted in up to 20% of patients with 2q37 deletions3). Septal defects are most common, and aortic coarctation has been described3). However, DCMP has not been reported in 2q37 deletion patients, and the relationship between DCMP and 2q37 deletion is not clear.
Our patient presented with short stature and was diagnosed as having partial GH deficiency. To date, there have been only 3 reports of GH deficiency in patients with 2q37 deletions6,8,9). Two cases were confirmed as 2q37 deletions6,9), and 1 case was confirmed as a distal 2p duplication and 2q deletion with severe short stature and pituitary hypoplasia8). All 3 reports noted a significant increase in growth rate after substitutive GH therapy, as shown in our patient.
Chromosome analysis confirms the diagnosis of 2q37 microdeletion syndrome in 80%–85% of affected individuals10). The largest telomeric deletion is about 10 Mb, while the smallest is around 3 to 4 Mb in the 2q37 chromosomal region. Deletion size does not appear to correlate well with phenotype11). The size of the 2q37 deletion was 8.3 Mb in our patient. The genes involved with 2q37.1q37.3 deletion are UTG1A4, UGT1A1, MLPH, COL6A3, PER2, TWIST2, HDAC4, NDUFA10, AGXT, KIF1A, PASK, and D2HGDH. The phenotypic implications of most of them remain unknown. Among the involved genes with a known phenotype, HDAC4 has notably been established as being responsible for brachymetaphalangia and intellectual disability12). In about 15%–20% of cases, the conventional karyotype is normal because of the small size of the deleted region10). Our patient showed a normal karyotype in the conventional chromosomal analysis; however, 2q37 microdeletion was identified by high-resolution cytogenetic analysis (i.e., array-CGH). Therefore, when patients are suspected of having a 2q37 deletion, high-resolution cytogenetic analysis is recommended3). Fluorescence in situ hybridization or array-CGH can be considered to confirm the diagnosis3).
It is important that chromosome 2q37 microdeletion be considered when patients have AHO features, especially in the presence of characteristic facial dysmorphism. We report a case of 2q37 deletion with DCMP, peripheral nerve palsy, and partial GH deficiency that responded well to GH therapy. When patients are suspected of having a 2q37 deletion, high-resolution cytogenetic analysis is recommended.