Introduction
Cushing syndrome (CS) is a disease resulting from glucocorticoid excess1). The incidence of endogenous CS is reportedly 2ŌĆō3 per million cases per year, and approximately 10% of cases are diagnosed during childhood 2,3). Endogenous CS is classified as adrenocorticotropin (ACTH)-dependent and ACTH-independent CS. ACTH-dependent Cushing disease, which comprises 70% of endogenous CS, is more common in adults and children of older age. ACTH-independent CS is more common in young children4) and is attributed to primary cortisol-producing adrenocortical lesions, which are adrenocortical tumors, adrenocortical carcinomas and primary adrenocortical hyperplasias. ACTH-independent CS is also associated with various diseases and genetic defects5,6).
We introduce an unusual case of a child with CS due to primary bilateral adrenal micronodular hyperplasia with spotty pigmentations on her conjunctivae and oral mucosa who had multiple osteoblastomas.
Case report
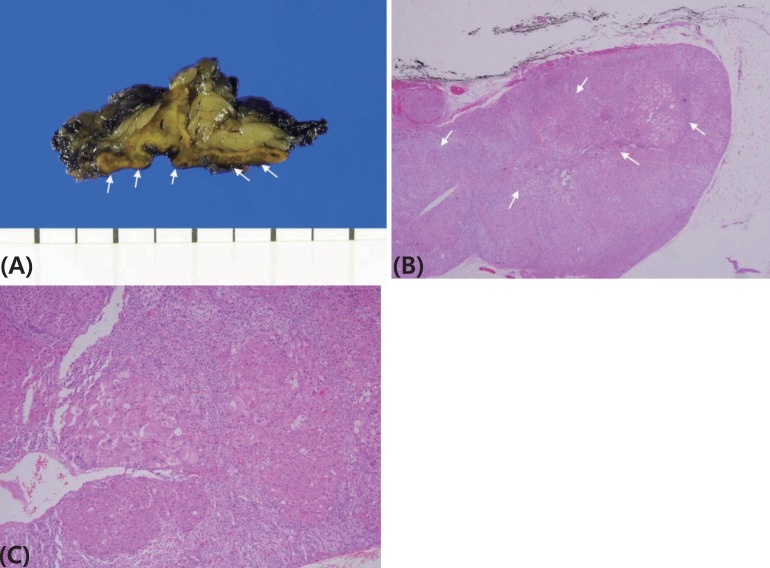
A 44-month-old girl was admitted who complained of excessive weight gain with growth failure and mood change. Her blood pressure was 150/80 mmHg, height was 96 cm (25th percentile), weighed 19.7 kg (>97th percentile) and her body mass index was 21.4 kg/m2 (>97th percentile). Plethoric moon face, hypertrichosis, and spotty pigmentation on her conjunctivae, lip and oral mucosa were observed. There was no specific medical history, including tumor or adrenal disease, in her family. Her urinary cortisol level was 966.2 ┬Ąg/m2/24 hr (reference range [RR], <70 ┬Ąg/m2/24 hr7)), and her serum cortisol level after a high dose dexamethasone suppression test was basal 34.6 ┬Ąg/dL (RR, 5ŌĆō23 ┬Ąg/dL), peak 32.9 ┬Ąg/dL (RR, <5 ┬Ąg/dL). The 24-hour urine cortisol levels of six-day Liddle's dexamethasone suppression test revealed following values; 1,380.0; 1,357.2; 1,064.4; 897.0; 944.0; 755.6 ┬Ąg/24 hr from day 1 to 6, respectively. The result was compatible to adrenal CS and no paradoxical increase was observed. Whereas her symptoms and signs of cortisol excess were very prominent, and her blood and urine cortisol levels were increased significantly, her computed tomography of the adrenal gland revealed 5-mm nodular thickening of the left adrenal gland and a nearly normal right adrenal gland (Fig. 1). She underwent laparoscopic left adrenalectomy at the age of 47 months. Because her symptoms of excess cortisol and increased urinary cortisol levels remained after left adrenalectomy, right adrenalectomy was performed at 58 months. The pathology of both adrenal glands revealed adrenocortical micronodular hyperplasia without apparent pigmentation, which was more compatible with micronodular adrenocortical disease (MAD) than primary pigmented adrenocortical disease (PPNAD) (Fig. 2).
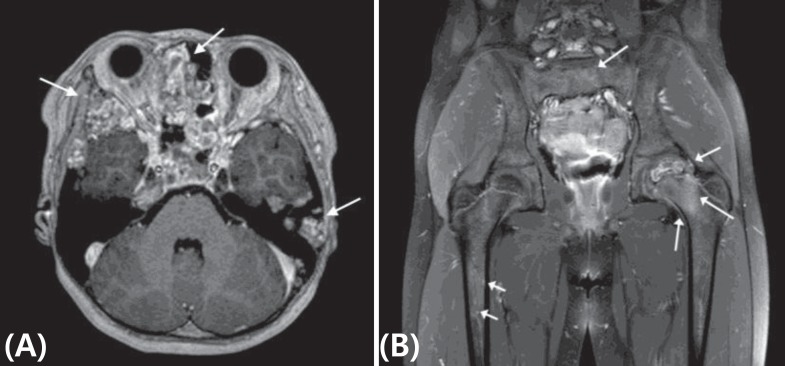
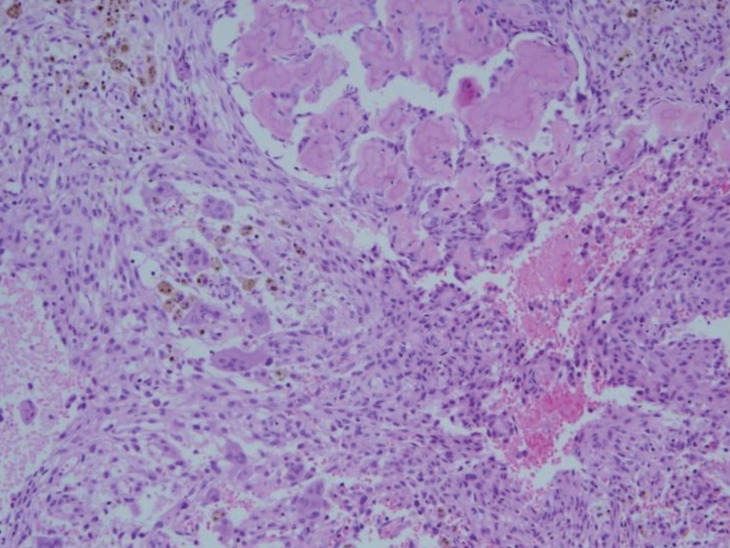
Because her skin pigmentation and pathology of both adrenal glands suggested the possibility of Carney complex, heart and thyroid gland ultrasonography were performed, which were all negative. No deletion or duplication was identified in protein kinase A regulatory subunit type 1A (PRKAR1A) gene analysis. However, at the age of 8 years, she complained of left hip pain, and her physical exam suspected a bone protrusion in the left temporal area. Magnetic resonance imaging (MRI) of her hip revealed overall polyostotic bone tumors involving the bilateral femurs, ilium and sacrum. Her brain MRI revealed multiple multilocular cystic bone lesions involving the clivus, bifrontal skull base, bilateral sphenoid, and left temporal bones (Fig. 3). The right temporal bone biopsy demonstrated osteoid producing neoplasms, consistent with osteoblastoma (Fig. 4). Because the bone pain had resolved and the skull, hip and leg lesions caused no functional problems, she was on conservative management with physiologic hydrocortisone and fludrocortisone replacement.
Discussion
Primary bilateral adrenal hyperplasias are rare diseases, the incidence of which is estimated to be up to 10% of ACTH-independent CS5). Bilateral adrenal hyperplasias are classified into 2 groups by the diameter of the associated nodules (macronodular, >1 cm; micronodular, Ōēż1 cm)5,6). While primary bilateral macronodular adrenocortical hyperplasia has a slight male predominance, micronodular hyperplasia has a slight female predominance and tends to arise at a younger age5,6). In cases of primary adrenal hyperplasia, medical treatments such as ketoconazole or mitotane which inhibits the biosynthesis of steroid can be used but has little effect. Eventually, unilateral (in case of adrenal adenoma or carcinoma) or bilateral adrenalectomy (in case of bilateral adrenal hyperplasia) is necessary4).
Micronodular adrenal hyperplasia is subdivided into a pigmented form 'PPNAD' and a limited or nonpigmented form 'MAD', although considerable morphological and genetic overlap is observed between the two groups6,8). In most of individuals with MAD, the adrenal glands had an overall normal size and multiple small yellow-to-dark brown nodules that were surrounded by a cortex with a uniform appearance. Microscopically, there was moderate diffuse cortical hyperplasia with mostly nonpigmented nodules9).
While MAD is usually an isolated disease, and the associated disease has not defined with MAD5,6), PPNAD typically occurs in the setting of Carney complex, which presents as cardiac myxoma and benign tumors in other body parts. The pathogenesis of Carney complex has been attributed predominantly to an inactivating germline mutation in protein kinase A regulatory-subunit type 1╬▒ (PRKAR1A) and in rare circumstance to alterations of the Carney complex type 2 (CNC2) gene loci. MAD cases have been associated with inactivating mutations in phosphodiesterase genes (PDE11A, PDE8B), and alterations of 2p12-p16 and 5q5,6,8,9).
Our patient demonstrated spotty skin, mucosal pigmentation and micronodular adrenal hyperplasia, which suggested Carney complex. However, a paradoxical increase in the 6-day Liddle's dexamethasone suppression test, which has been described in patients with PPNAD, did not present, and PRKAR1A mutations were negative in this case. Moreover, the pathology revealed neither pigmentation nor internodular atrophy of the adrenal cortex, which was more favorable to MAD. Bone tumors associated with Carney complex are known as osteochondromyxomas. They usually present without pain and are detected during the imaging evaluation of mass side effects, and pathology reveals osteolytic lesions composed of myxoid matrix10,11). However, in our case, the first presenting bone tumor symptoms were pain and bone protrusion, and biopsy revealed osteoid producing neoplasm, consistent with osteoblastoma. This suggests that the bone tumor is a different entity in this case rather than related bone tumors such as Carney complex. Since osteoblastomas are a benign bone forming tumor, surgical curettage or resection are needed if symptoms are present or bone destructions are observed12). About 26.6% of the patients are treated nonsurgically. And among the patients who underwent surgical procedure, recurrence rate after the surgery is 24.4%12,13). But in our case, the pain was resolved and no other functional problem occurred, so conservative therapy was done without any surgery.
While MAD is usually an isolated case, our case is the first reported case that presented with spotty pigmentation and multiple osteoblastomas. Judging by the above-mentioned information, we can cautiously conclude that this patient might have a proliferation malfunction or a mutation in a tumor suppressor gene. Although we could not find the exact gene, we should carefully screen due to probabilities of other neoplasms.
Various symptoms and clinical outcomes can result from CS, which makes the diagnosis difficult and subtle. This case highlights the importance of verifying the etiology of CS as well as careful follow-up and screening for associated diseases.