Introduction
Prader-Willi syndrome (PWS; OMIM #176270) is an imprinted neurobehavioral condition affecting many organ systems and occurs due to the absence of expression of a cluster of paternally expressed genes located at 15q11-q131). PWS is characterized by 2 phases in terms of clinical features. In infancy there is a failure to thrive, muscular hypotonia, genital hypoplasia, respiratory problems, and feeding difficulties2). From as early as 2 years of age, an altered phenotype becomes apparent with evidence of mild developmental delay and learning disabilities and the onset of severe overeating behavior resulting from an abnormal satiety response to food intake3). Other later-phase phenotypic characteristics include growth hormone deficiency, short stature, small hands and feet and significant behavioral problems1,2). Recent epidemiological study estimates an incidence of 1 in 25,000 births and a population prevalence of 1 in 50,0004). Paternally expressed genes are particularly important in hypothalamic development, as indicated by the hypothalamic accumulation of androgenetic (duplicated paternal genome) cells in chimeric mouse embryos5,6). Paternal de novo deletions of the 15q-q13 region account for about 70% of PWS. Most of the remaining cases have uniparental maternal disomy (UPD) for chromosome 15. The PWS region includes a few protein-coding genes and multiple paternally expressed noncoding RNAs, several of which were previously suggested to regulate alternative splicing7,8). Several imprinted genes or transcripts have been mapped to15q11-q13, most with only paternal expression, including SNURFŌĆōSNRPN, several clusters of small nucleolar RNAs (snoRNAs), NDN, MKRN3, NPAP1, and MAGEL29). The noncoding RNAs are highly expressed in the brain and function through modification of ribosomal RNAs10). Nevertheless, the functions of the vast majority of genes residing in the PWS region remain to be determined. The syndrome has a clinical overlap with other diseases, which makes it difficult to accurately diagnose. The challenge for clinicians is not only to differentiate more clearly between PWS and the various Prader-Will-Like syndrome (PWLS) on a clinical level but also to provide conclusive genetic explanations for these phenotypes to provide accurate genetic counseling and treatment. The absence of a correct diagnosis may worsen the prognosis of these individuals due to the endocrine-metabolic malfunctioning associated with the PWS. Therefore, an accurate chromosomal investigation is necessary to differentiate classical PWS from the PWLS. This review aims to provide an overview of current knowledge relating to the genetics of PWS and PWLS.
Molecular and genetic basis of PWS
1. Structure and genes in the 15q11-q13 region
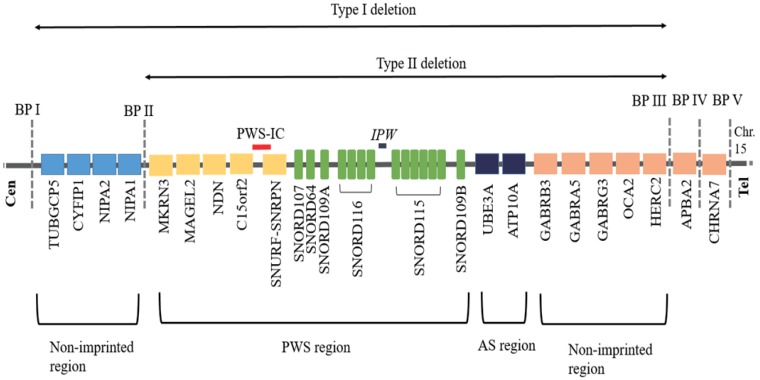
The 15q11.2-q13 region can be roughly divided into 4 distinct regions that are delineated by 3 common deletion breakpoints11), which lie within segmental duplications12) (Fig. 1). First, proximal nonimprinted region between the 2 common proximal breakpoints (BP I and BP II) containing four bi-parentally expressed genes, NIPA1, NIPA2, CYF1P1, and TUBGCP513). Second, the PWS domain contains five paternally expressed protein-coding genes (MKRN3, MAGEL2, NDN, snoRNAs, and SNRPN-SNURF, C15orf2) and several antisense transcripts (including the antisense transcript to UBE3A)9,14). Third, the Angelman syndrome (AS) domain containing the preferentially maternally expressed genes (MEGs) (ATP10A and UBE3). Fourth, a distal nonimprinted region containing a cluster of three gamma-aminobutyric acid receptor genes, the gene for oculocutaneous albinism type 2 (OCA2), HERC2, and the common distal breakpoint (BP III).
The genomic and epigenetic changes causing PWS all lead to a loss of expression of the normally paternally expressed genes on chromosome 15q11.2-q1315). Absence of the paternally inherited copy of these genes, or failure to express them, causes total absence of expression for those genes in the affected individual because the maternal contribution for these genes has been programmed by epigenetic factors to be silenced16). The 15q11.2-q13 region is highly vulnerable to structural rearrangements, such as deletions, duplications, supernumerary marker chromosomes, and translocations due to presence of low-copy repeats (LCRs) in the region17,18). The exact function of each of the genes in determining the PWS phenotype remains to be elucidated, although possible insight has been gained by work with mouse models by multiple investigators15).
2. Allelic variants related to PWS
PWS is a contiguous gene disorder, as studies thus far indicate that the complete phenotype is due to the loss of expression of several genes15). The search for candidate genes contributing to specific phenotypic components of PWS has been extensively performed.
1) SNURF-SNRPN gene
Central to the PWS region is the SNURF-SNRPN gene which is an extremely complex gene locus that spans, 465 kb, with 148 possible exons that undergo alternative splicing4,19). It is a bicistronic gene encoding two different proteins15). Exons 4ŌĆō10 were described first and encode the protein SmN, which is a spliceosomal protein involved in mRNA splicing20). SNURF is encoded by exons 1ŌĆō3, which produces a polypeptide of unknown function21). At the 5' end of the SNURF-SNRPN gene is a CpG island encompassing the promoter, exon 1, and intron 115). Imprinting occurs partly through parent-of-origin allele-specific methylation of CpG residues, which is established either during or after gametogenesis and maintained throughout embryogenesis22). This is a differentially methylated region, which is unmethylated on the paternally inherited expressed allele and methylated on the maternally inherited repressed allele20). The SNRPN minimal promotor region includes 71 bp of upstream sequence and the first 51 bp of SNURF-SNRPN exon 123,24) and the integrity of the SNRPN minimal promoter region/exon1 region appears to be essential part of the imprinting center (IC) in the PWS chromosomal region in both mice and humans, and upstream sequences are critical for the correct function of either mechanism25,26). The SNURF-SNRPN gene also serves as the host for the six snoRNA genes which are regulated by the expression of SNURF-SNRPN and do not encode proteins19,27,28). Changes in the noncoding regions can cause genetic disease by altering gene expression. Wu et al.29) showed that mutations upstream of SNRPN/exon1 caused lack of methylation in the maternal SNRPN promoter and activation of MEGs including a rescue from the lethality and growth retardation normally shown by the PWS mouse. Maina et al.23) suggested that changes in the maternal SNRPN minimal promotor region may ameliorate some of the more severe symptoms of the disease shown by 9 PWS patients with atypical genetics.
2) snoRNA genes
The snoRNA genes located in the large SNURF-SNRPN transcripts present not only in single copy (SNORD64, SNORD107, SNORD108, SNORD109A, and SNORD109B) and but also in the two snoRNA gene clusters (SNORD115 and SNORD116)9,15). It is thought that the snoRNAs may target cellular mRNAs for methylation or alternative splicing because the snoRNAs in the PWS region lack the usual rRNA complementarity and that each snoRNA gene might have multiple targets19,27). The snoRNA gene may be responsible for at least several features of PWS on the basis of 6 patients with balanced translocations affecting the SNURF-SNRPN locus, who were described to have typical PWS or a PWLS phenotype30,31,32). A "key" region to explain much of the PWS phenotype has been narrowed to the SNORD116
snoRNA gene cluster33). Mice lacking the SNORD116 orthologue display a partial PWS phenotype34). The SNORD116 has been shown to be highly conserved in rodents, and Gallagher et al.28) showed that a 121-kb region thought to be critical in PWS contained both SNORD116 and SNORD109, suggesting that these genes may play a major role in the PWS phenotype. Up to date, most reported clinical cases of limited deletion of the SNORD116 cluster associated with PWS have also involved adjacent genes: SNURF-SNRPN or SNORD11534,35). But, a crucial role for the SNORD115 locus was eliminated by an AS family with a familial microdeletion that included the entire SNORD115 gene cluster and the UBE3A locus36). There have been three separate reports of three different individuals with overlapping microdeletions (175ŌĆō236 kb) that all encompass the SNORD116 gene cluster34,35). All three have multiple clinical features typical of PWS including neonatal hypotonia, infantile feeding problems, rapid weight gain by 2 years of age, hyperphagia, hypogonadism, developmental delay/intellectual disability, and speech and behavioral problems15). However, these three individuals also have features not typical of classical PWS, including tall stature, macrocephaly, lack of a "PWS facial gestalt," and atypical hand features of PWS. More recently, Bieth et al.37) reported the first case of a patient with the highly typical features of PWS who presented a restricted deletion of the SNORD116 region which did not affect the expression of SNURF-SNRPN and did not delete any portion of the SNORD115 locus. This finding in a human case might suggest that a lack of the paternal SNORD116 gene cluster has a determinant role in the pathogenesis of PWS.
3) MAGEL2 gene
The MAGEL2 is located adjacent to NDN in the human and mouse, with highest expression in mouse at late developmental stages and in the hypothalamus and other brain regions and considered to be a candidate gene for the eating disorder of PWS38,39,40). Wevrick et al.41,42,43) have reported that Magel2-null mice have selected biological findings similar to PWS in humans, including neonatal growth retardation, excessive weight gain after weaning, impaired hypothalamic regulation and reduced fertility. Recently, Schaaf et al.14) reported 2 patients with point mutations in the imprinted MAGEL2 gene in the 15q11-q13 domain causing classic PWS, suggesting that that MAGEL2 loss of function can contribute to several aspects of the PWS phenotype.
4) NDN gene
Among the imprinted candidate genes for PWS, the gene NDN encoding the MAGE family NECDIN protein proposed to act as a neuronal growth suppressor and antiapoptotic protein in postmitotic44). A mouse Ndn knockout model has been reported with similar defects to individuals with PWS45) and mouse Ndn mRNA is expressed predominantly in a subset of postmitotic neurons, with highest levels in the hypothalamus and several other brain regions at late embryonic and early postnatal stages, as well as other tissues46).
5) MKRN3 gene
The MKRN3 encodes the makorin ring finger protein 3 and the MKRN3 differential allele expression occurs through silencing of the MKRN3 maternal allele, which is associated with 5' CpG island methylation47). The functional and physiological relevance of MKRN3 is not well known and despite its location in the PWS critical region, its role in this syndrome is also unclear. Recently, it was investigated if central precocious puberty (CPP) could arise from loss of MKRN3 expression by the paternal allele due to a de novo deletion, maternal UPD or an imprinting defect, mechanisms recognized in the pathogenesis of the PWS48). Particularly, a girl with a paternal deletion in MKRN3, MAGEL2 and NDN genes, who had few features of the PWS, was diagnosed as CPP49). In addition, few cases with the PWS and CPP have been reported50). Potentially, human genomic sequence and global methylation analyses in PWS patients with CPP could establish epigenetic alterations in the pathogenesis of the disorder.
6) IPW
Many long noncoding RNAs (lncRNAs) interact with chromatin-modifying proteins owing to their secondary structure and can recruit chromatin-modifying complexes to specific genomic regions. IPW located within the critical PWS-associated region on chromosome 15 is one of several lncRNAs associated with an imprinted locus, which is considered to be an RNA transcript only, because it does not encode a protein1). Stelzer et al.51) demonstrate that a paternally expressed lncRNA known as IPW has a role in modulating the expression of MEGs and identified a critical role for IPW in modulating the expression of MEGs in trans, which has important implications for the understanding of imprinted gene networks. Loss of imprinted genes in the PWS locus thus leads to an effect in trans of increased expression of imprinted genes in the DLK1-DIO3 locus on chromosome 14, suggesting that there might be cross-talk between imprinted loci.
7) Nonimprinted genes (NIPA1, NIPA2, CYFIP1, and GCP5)
It is unclear whether the four, nonimprinted genes (NIPA1, NIPA2, CYFIP1, and GCP5) localized to the interval between BP I and BP II, contribute towards the PWS phenotype44).
3. Molecular classes of PWS
There are 3 main classes of chromosomal abnormalities that lead to PWS: deletion on 15q11-q13, maternal UPD of chromosome 15, or a defect in the IC on 15q11-q13, although gene mutation (<0.1%) and balanced translocation (0.1%) can also be found52,53).
1) Microdeletion of the chromosome region 15q11-q13 deletion
Most patients with PWS result from an interstitial microdeletion of the paternally inherited 15q11.2-q13 region. Deletions occur in about 65%ŌĆō75% of the patients with PWS and AS. Deletions in PWS and AS are subdivided into 2 main subgroups (types I and II) and the BPs are flanked by LCRs in the 3 BPs12). Two common classes of deletions of the PWS/AS critical region have been described; type I (40%), approximately 6 Mb in size between BP I and BP III and type II (60%), spanning 5.3 Mb between BP II and BP III54). Individuals with deletion of type I show a more severe phenotype than type II11,12,55). Both types I and II deletions are almost always de novo events56). These recurrent common interstitial deletions measure approximately 5ŌĆō6 Mb in size and are due to the presence of multiple copies of tandemly repeated sequences at the common breakpoints (BP I, BP II, and BP III) flanking the deleted region15). These LCRs sequences stretch for approximately 250ŌĆō400 kb and can cause nonhomologous pairing and aberrant recombination of the 15q11.2-q13 region during meiosis, leading to deletions, duplications, triplications, and inverted dup (15)57). In addition, approximately 8% of those with a deletion have a unique or atypical sized deletion (i.e., not type I or II) from a variety of etiologies, including an unbalanced translocation56). A deletion that is smaller or larger than typically seen in PWS may affect the phenotype rare and have been important for the delineation of genotype-phenotype correlation9).
2) Uniparental disomy of chromosome 15
Maternal UPD 15 is the situation in which there are 2 chromosomes 15 from the mother and none from the father58). This accounts for approximately 20%ŌĆō30% of individuals with PWS. Maternal UPD has been shown to be associated with advanced maternal age59,60). Trisomy associated with Robertsonian translocations may resolve to disomy through loss of a chromosome and would result in UPD in 50% of cases15). UPD can be associated with small supernumerary chromosome 15 markers, and both maternal and paternal UPD 15 have been identified from this situation, although maternal is more common61). The parental origin of these small markers is frequently unknown due to the small size and lack of unique genetic material. It has been estimated that approximately 5% of small supernumerary markers are associated with UPD62).
3) Imprinting defect
This molecular class affects the imprinting process on the paternally inherited chromosome 15 and accounts for approximately 1%ŌĆō3% of individuals with PWS15). Most IDs result from epigenetic causes (epimutations) and demonstrate a maternal-only DNA methylation pattern despite the presence of both parental alleles (i.e., biparental inheritance)15). DNA sequence changes are not found in these epimutations, and they are thought to be random errors in the imprinting process or in early embryogenesis in the rare cases of somatic mosaicism16). However, approximately 15% of individuals with an ID are found to have a very small deletion (7.5 to >100 kb) in the PWS IC region located at the 5' end of the SNRPN gene and promoter (i.e., IC deletion)63). Of these, about half have been inherited from an unaffected father with the IC deletion on his maternally inherited chromosome 1515). The other half are de novo IC deletions on the paternally inherited 15 that occur during spermatogenesis in the father or after fertilization59,64).
Genotype-phenotype correlations
There are no features known to occur exclusively in individuals with one of the genetic classes15). However, there are some statistical differences in the frequency or severity of some features between the 2 largest classes (deletion 15q11.2-q13 and UPD)15). Postterm delivery is more common with UPD65). Individuals with UPD are less likely to have the typical characteristic facial appearance59,60), or skill with jigsaw puzzles66). Patients with deletions have a higher frequency of hypopigmentation of skin, hair and eyes due to loss of expression of the nonimprinted P gene that is involved in oculocutaneous albinism59,60). In most studies, those with UPD have a little higher verbal IQ and milder behavior problems67). Interestingly, psychosis and autism spectrum disorders (ASD) are almost shown to PWS adults with UPD rather than deletions68). Recent studies suggest that as many as 62% of those with UPD develop atypical psychosis compared with 16% of those with a deletion69). PWS subjects with IC mutations appear to have a classical PWS phenotype and might have a similar increased predisposition to psychosis as UPD70). Torrado et al.71) reported that patients with a deletion type had a higher frequency of need for special feeding techniques, sleep disturbance, hypopigmentation, and speech articulation defects. Several studies have investigated phenotypic characteristics between PWS individuals with type I versus type II deletions, but there has been a lack of consensus among the different studies. For example, Butler et al.72) reported 12 patients with type I deletion showed worse adaptive behavior, more severe compulsive behavior and more impairments in reading, math skills and visual perception than 14 patients with type II deletion. On the other hands, Milner et al.73) did not find any significant phenotypic differences between the 2 main deletion subtypes (type I, n=14; type II, n=32). As the genotypeŌĆōphenotype relationships become clearer, it will be clinically important to readily subtype the deletion class56).
Clinical manifestations and molecular genetics of PWLS
PWLS share features of the PWS phenotype, however the genetic basis of these rare disorders differs. The implication is that the gene functions disrupted in PWLS are likely to lie in genetic pathways that are important for the development of PWS phenotypes. The main clinical manifestations of PWLS were reported including hypotonia, obesity, short extremities, and delayed development74). However, PWLS phenotype is clinically and genetically heterogeneous. Recently, several patients with PWLS (ASD, intellectual disability, and a varying degree of clinical and behavioral features of PWS) were reported in whom four different de novo heterozygous truncating mutations in the MAGEL2
gene were identified which occurred on the paternal allele, suggesting that MAGEL2 is a novel gene causing complex ASD, and MAGEL2 loss of function can contribute to several aspects of the PWS phenotype14). Rocha, et al.52) reviewed a total of 117 PWLS patients. Of these patients, 44 had their final genetic diagnosis established. Their most frequent symptoms were obesity (84%), hyperphagia (72.7%), mental disability (54.5%), psychomotor delay (50%), and hypotonia (43.18%). It is important to recognize that signs and symptoms of PWS could also be found in patients who show other types of chromosomal abnormalities. For example, chromosomal abnormaliticoes such as chromosome 14 maternal uniparental disomy75), 1p36 monosomy76), deletion of 6q74,77,78), 2pter deletion79), 10q26 deletion80), paracentric inversion (X)(q26q28)81), 12q subtelomere deletions82), Xq27-qter disomy, deletion of 3p26.322), fragile X83), fragile X with 47,XYY84), duplication of X(q21.1-q21.31)85), and a duplication of Xq23-q25 duplication86) could be associated with the PWLS phenotype. Genes that are outside of the PWS region result in PWLS phenotypes, and are thus implicated in important pathways in PWS pathobiology that might require additional research. For example, mutations in SIM1, leptin receptor (LEPR), pro-opiomelanocortin (POMC), melanocortin 4 receptor (MC4R), and more recently, POU3F2 have all been related to severe obesity which suggests a convergence onto the leptin-melanocortin pathway in association with oxytocin87,88). Among them, the SIM1 gene, mapping to the common 6q16.2deletion region, has been proposed as a candidate for the obesity observed in all these subjects. The 4.1 Mb critical region for PWS includes SIM1 but also 11 other genes or gene predictions, and the specific role of SIM1 haploinsufficiency in the development of PWS has not definitively been established77). Bonnefond et al.89) sequenced SIM1 gene in 44 children with PWLS features, 198 children with severe early-onset obesity, 568 morbidly obese adults, and 383 controls. They identified 4 rare variants (p.I128T, p.Q152E, p.R581G, and p.T714A) in 4 children with PWLS features (including severe obesity) and 4 other rare variants (p.T46R, p.E62K, p.H323Y, and p.D740H) in 7 morbidly obese adults. Three mutations showed strong loss-of-function effects (p.T46R, p.H323Y, and p.T714A) and were associated with high intra-family risk for obesity, while the variants with mild or no effects on SIM1 activity were not associated with obesity within families. It suggested a firm link between SIM1 loss of function and severe obesity associated with, or independent of, PWS-related clinical features53). Al Ageeli et al.90) reported case with SMC15 of paternal origin and PWLS features: motor and intellectual delay, autism, auto-aggressivity, attention deficit and excessive eating, obesity, and facial dysmorphism (round face, deep set eyes, narrow palpebral fissures, epicanthus, and upturned nose with broad nasal bridge). A comparative study investigating the prevalence and severity of obsessive-compulsive symptoms (OCS) in PWS and PWLS showed that PWS patients suffered a higher incidence of OCS and more severe symptoms than their PWLS counterparts91).
Clinical diagnosis and diagnostic testing
Even though consensus clinical diagnostic criteria for PWS were established in 1993, 17% of 90 patients with a molecular PWS diagnosis did not fulfill the consensus clinical diagnostic criteria92). Therefore, they suggested new criteria to prompt DNA testing for PWS (cognitive impairment, excessive eating, central obesity, and hypothalamic hypogonadism). DNA methylation analysis is the most efficient way to start the genetic workup if PWS is suspected clinically (Fig. 2). DNA methylation analysis is the only technique that will diagnose PWS in all 3 molecular classes and differentiate PWS from AS in deletion cases, and a methylation analysis consistent with PWS is sufficient for clinical diagnosis16,20). The most robust, and now most widely used, assay targets the 5' CpG island of the SNURF-SNRPN locus, and it will correctly diagnose PWS in more than 99% of cases20). Although, the methylation analysis is the gold standard technique for detecting PWS, it cannot distinguish the molecular class (i.e., deletion, UPD, or ID). Therefore, cytogenetic analysis should also be performed, not only to look for a 15q11-q13 deletion, but to find other chromosomal abnormalities52). For genetic counseling purposes, a chromosomal analysis is also recommended in the proband to discern an interstitial de novo deletion from a balanced or unbalanced chromosomal rearrangement involving the 15q11.2 region. Traditionally, deletions of 15q11.2-q13 have been diagnosed with Fluorescence in situ hybridization (FISH) with the SNRPN probe15). With the increasing use of chromosomal microarray (CMA) in clinical genetics, it is possible that arrays may replace FISH analysis for the identification of deletions in PWS and AS15). CMA will precisely report not only the deletion size, but also additional chromosomal abnormalities elsewhere in the genome, which is anticipated to become increasingly important for genotype-phenotype correlations in the future44). However, CMA will not identify the rare chromosomal rearrangements (translocations and inversions) involving proximal 15 which are detectable by simultaneous karyotype and FISH analysis and are important in recurrence risk determination.
If DNA methylation is positive for PWS (i.e., maternal only imprint), but no deletion is found, the next step is to distinguish between maternal UPD and an ID15). This is accomplished by using DNA polymorphism analysis of chromosome 15 loci on the proband's and parents' DNA, which can diagnose UPD in some cases but not all93). If the family polymorphism study reveals that the proband has biparental inheritance of chromosome 15 loci (rather than maternal UPD), then the molecular class is presumed to be an ID15). It is then important to determine whether the ID is due to an epimutation (low recurrence risk) or a small deletion in the IC, as in the later situation the recurrence risk can be as high as 50% if the father also has an IC deletion15). Approximately 15% of individuals with PWS due to ID have an IC deletion and approximately 50% of these are familial mutations78). Testing for IC deletion can be done by sequence analysis at the smallest region of overlap for the PWS IC, which is a region of approximately 4.3 kb63,78), or by the recently developed methylation-specific multiplex-ligation probe amplification (MS-MLPA) assay. Some laboratories, particularly in Europe, now begin testing for PWS by using the MS-MLPA analysis (rather than single locus DNA methylation analysis at 5'SNRPN), with a detection rate of more than 99%15). Compared to traditional DNA methylation, the advantage of MS-MLPA is that the MS-MLPA will investigate 5 distinct differentially methylated sites rather than just one locus and will give information on dosing in the 15q11.2 region15). The latest kit has particularly dense probe coverage for dosing and DNA methylation analysis in the PWS "critical region" between the PWS IC and SNORD11615). In addition, the MS-MLPA technique is much more labor/cost-effective than CMA analysis, although CMA provides more precise information regarding the extent of the deletion44). Therefore, MS-MLPA analysis might be considered as the first testing when suspecting AS or PWS as a possible diagnosis, especially since important genotypeŌĆōphenotype correlations will likely be forthcoming44). However, MS-MLPA will also not detect chromosomal rearrangements (inversions and translocations) involving proximal 15, which are detectable by simultaneous karyotype and FISH analysis15).
Genetic counseling
Understanding the specific genetic etiology in individuals with PWS is vital for the appropriate genetic counseling of affected families15). Estimation of recurrence risk is dependent on the genetic defect causing PWS. Deletion 15q11.2ŌĆōq13 is sporadic (recurrence risk<1%) except in the rare cases where a chromosomal balanced rearrangement (translocation or inversion) is present in the father94). In these cases, there is a theoretical recurrence risk as high as 25%ŌĆō50%. In addition, a scenario with a risk of 100% is very unlikely but theoretically possible (i.e., a mother with a 15/15 Robertsonian translocation). Although chromosome rearrangements are the most infrequent genetic cause (<1%), it is important to analyze the karyotype of patients suspected of PWS to identify chromosome 15 rearrangements, plus other chromosomal anomalies because the karyotype and FISH analyses carried out in the affected child could give enough information to suspect whether the deletion comes from a chromosome rearrangement95,96). Small supernumerary marker chromosomes have been reported in ~0.3% of mentally retarded patients97), and in most cases, the small supernumerary marker chromosomes could be derived from chromosome 15, resulting in a UPD61). Only in these cases are studies on fathers recommended to offer a thorough genetic counseling98). Maternal UPD 15 is typically de novo (recurrence <1%) except if a Robertsonian translocation is present in either parent. If the patient chromosomal analysis is normal, it must be expected that the maternal UPD be sporadic98). Taking together, there are two important considerations. First, the patient chromosomal analysis is also important to identify the presence of small supernumerary marker that could explain some maternal UPD cases. Second, if a Robertsonian translocation is identified in the patient karyotype and suspected as the origin of a maternal UPD, then it is the mother karyotype that must be performed instead of the father one97). Approximately 15% of those with an imprinting defect have a microdeletion in the IC; this can be familial and has a 50% recurrence risk when it is97). Therefore, fathers of children with an IC deletion should have DNA methylation and dosing analysis (or sequence analysis) to determine whether they carry the IC deletion15). However, the greater proportion of those with an imprinting defect have an epigenetic mutation and the recurrence risk is <1% for this group98).
Conclusions
A wide range of genetic conditions are associated with appetite control, body composition, growth, reproduction, learning disability, psychosis and other behavioral problems. As yet, many outstanding questions remains to be answered about these condition although much is being learnt relating to the important contribution of specific genes, and their coded proteins. The genetic complexity of the PWS chromosomal region, with multiple imprinted genes, alternative splice variants, gene duplications and variant copies, and the mechanisms of imprinting itself, are matched by the wide variety of phenotypes that involve multiple organ systems. Updated information regarding the genetics and phenotypic characterization of individuals with PWS and PWLS is important for all physicians and will be helpful in recognizing patients that may benefit from further investigation and genetic screening and anticipating complications associated with this rare obesity-related disorder.