Introduction
The term congenital adrenal hyperplasia (CAH) covers a group of autosomal recessive disorders caused by a deficiency of one of the enzymes required for steroid biosynthesis in the adrenal gland1). 21-Hydroxylase deficiency (21OHD) is the most common type of CAH, accounting for approximately 95% of cases, and is caused by mutations in the CYP21A2 gene2). 21OHD is clinically divided into classic forms, including salt-wasting (SW) and simple virilizing (SV) forms, and nonclassic forms of disease according to disease severity3). Most patients with classic CAH present with SW crisis or ambiguous genitalia in the neonatal period. In contrast, boys do not show ambiguous genitalia and therefore cannot be diagnosed without neonatal screening4). Therefore, males with CAH have a higher risk of adrenal crisis in the neonatal period if they were the SW form of 21OHD5).
The level of 17α-hydroxyprogesterone (17OHP) is used as a marker for 21OHD, as CAH due to 21OHD can be detected in newborn screening programs by measuring the amount of 17OHP in dried blood spots6). Newborn screening for CAH is now performed in many countries to prevent SW crises in the neonatal period, to prevent male sex assignment in affected females, and to reduce long-term morbidities, such as short stature, gender confusion, and psychosexual disturbances6).
It is critical to diagnose 21OHD in a timely manner and treat classic and nonclassic CAH to prevent adrenal crises and hyperandrogenism. This review will describe the current knowledge regarding newborn screening and biochemical and molecular genetic diagnoses of CAH due to 21OHD.
Newborn screening for CAH due to 21OHD
The goal of newborn screening for CAH is to detect the SW adrenal crisis; to prevent shock, brain damage, or death and begin presymptomatic treatment; and to prevent or shorten the period of incorrect gender assignment in affected females7,8). The benefit of newborn screening is to identify boys with the SW form of CAH, as they do not have ambiguous genitalia at birth9). For effective newborn screening, a careful clinical follow-up is critical to confirm diagnosis and early treatment4). Neonatal screening tests were first performed for congenital hypothyroidism and phenylketonuria in Korea in 1997. The measurement of 17OHP to screen for CAH has been included in the newborn screening program in Korea since 200610). The incidence of CAH due to 21OHD in the Korean population detected by newborn screening is 1 in 22,700 (http://helpline.cdc.go.kr/).
The biochemical marker for the diagnosis of 21OHD is elevated 17OHP, the main substrate for the enzyme lying just upstream of the block4,11). Using filter paper cards, newborn screening programs measure 17OHP in dried blood spots obtained via a heel puncture performed between 2 and 4 days after birth1,12). Because the circadian rhythm is influenced by cortisol13), an early morning 17OHP level is appropriate for screening4). The 17OHP level in normal newborns is <1 ng/mL (3 nmol/L)1). After the neonatal period, a cutoff value of an early morning 17OHP <0.8 ng/mL (2.5 nmol/L) in children and <2 ng/mL (6.0 nmol/L) in adults has been suggested to exclude CAH14). A random blood sample with 17-OHP >100 ng/mL (300 nmol/L) measured by radioimmunoassay (RIA) is diagnostic of classic 21OHD, while 1–100 ng/mL (30–300 nmol/L) in adults indicates nonclassic 21OHD2). In menstruating women, the samples for 17OHP should be measured in the follicular phase, as the 17OHP level usually increases in the luteal phase in about half of normal females with a level of >2 ng/mL (6.0 nmol/L)15).
Various techniques for measuring 17OHP are available, including RIAs, enzyme-linked immunosorbent assays, and time-resolved fluoroimmunoassays. The RIA was the first method used, but the dissociation-enhanced, lanthanide fluorescence immunoassay is now more common6). Each method has advantages and disadvantages in terms of specificity and sensitivity.
One of the disadvantages of newborn screening for 21OHD is the considerably high false positive rate, leading to high rate of repeated tests that cause patient distress and result in significant costs for follow-up. The false positive results are attributed to several factors, such as prematurity, sickness, stress, and low specificity of antibodies for 17OHP due to the cross-reactivity with 17-hydroxypregnenolone and immature adrenal steroid production4,16,17,18,19).
Recent advances in biochemical analysis of adrenal steroid profiles
Primary screening results for CAH using a 17OHP assay must be confirmed by second tier tests, including biochemical or molecular genetic analyses6). In addition, because of the relatively high false positive rate of the first tier screening using immunoassays, there is a need for highly specific 17OHP screening methods to select patients for further diagnostic approaches.
Tandem mass spectrometry (MS/MS) has been recently developed to improve the positive predictive value and is more specific than the immunoassays20). Second-tier strategies using MS/MS measure a ratio of 17OHP plus androstenedione to cortisol20,21). However, they are time-consuming to carry out and are not appropriate as first-tier screening methods20).
Liquid chromatography linked with MS/MS (LC-MS/MS) is a revolutionary method to measure steroid hormones in various body fluids to improve sensitivity and specificity in highly automated systems22). LC-MS/MS allows for rapid, targeted steroid hormone analysis of multiple analytes23). It is currently the technique of choice for confirming CAH, as molecular genetic analysis is time-consuming and expensive24). The issues of cross-reactivity and specificity can be alleviated by the use of LC-MS/MS22). However, the problem of false-positive results in premature or stressed newborns cannot be completely overcome simply by determining 17OHP alone. LC-MS/MS can measure 17OHP levels as well as other compounds, such as androstenedione, 11-deoxycortisol, 21-deoxycortisol, and cortisol25,26). The use of analyte ratios (precursor/product), such as (17OHP+21-deoxycortisol)/cortisol, can effectively decrease the possibility of the false positive results in neonates with high 17OHP levels due to stress, sickness, or prematurity6,27). In nonclassic CAH, analyte levels are only slightly different compared to those in healthy newborns, but analyte ratios demonstrate more pronounced alterations than analytes themselves6). Therefore, the specificity and sensitivity of LC-MS/MS and the use of analyte ratios are expected to facilitate the diagnosis of mild, nonclassic CAH.
A urine steroid profile can be performed using a spot urine sample and is an additional CAH diagnostic tool that helps to differentiate between the different forms of CAH28). Gas chromatography-mass spectrometry (GC-MS) is used for urine steroid profile analysis, which is a helpful noninvasive diagnostic test used for confirmatory diagnosis29). As markers for 21OHD, urinary 17OHP metabolites, such as 17α-hydroxypregnanolone, pregnanetriol, and 15β,17α-dihydroxypregnanolone, and urinary 21-deoxycortisol metabolites, such as pregnanetriolone, have been studied using GC-MS25,30). However, both mass spectrometric techniques are complementary tools for diagnosing CAH, and further study is still required to improve reproducibility between laboratories23).
Indication of cosyntropin stimulation test
Marked elevation of 17OHP levels is characteristic of classic 21OHD. However, equivocal values of 17OHP cannot distinguish nonclassic CAH from heterozygous carriers31) and require dynamic testing with corticotropin (cosyntropin) stimulation2). This test is the gold standard for diagnosing nonclassic CAH4). The cosyntropin stimulation test is performed by intravenously injecting cosyntropin at a dose of 0.25 mg and measuring baseline and stimulated levels of 17OHP1). Blood samples are obtained at baseline and 60 minutes after the administration of cosyntropin. Cortisol levels should also be measured at baseline and 60 minutes to measure the cortisol level. A basal 17OHP >5 ng/mL (15 nmol/L) and/or peak 17OHP >10 ng/mL (30 nmol/L) traditionally indicates nonclassic CAH32).
Genetics of 21OHD
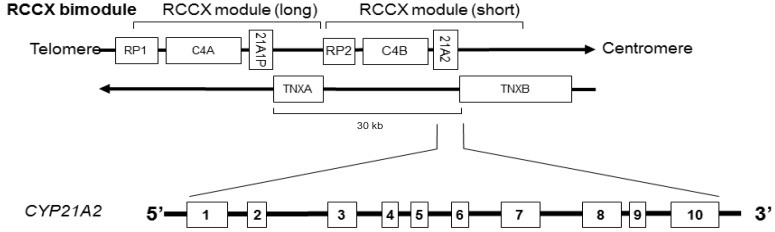
The CYP21A2 gene encodes 21-hydroxylase and requires electrons transferred from NADPH via the electron donor enzyme P450 oxidoreductase. This gene is located in the HLA class III region between the HLA-B and HLA-DR chromosome 6p21.31). This is a highly complicated region including a highly homologous pseudogene, CYP21A1P1). The functional gene (CYP21A2) and a nonfunctional pseudogene (CYP21A1P) are located closely adjacent to each other in tandem arrangement with the C4A and C4B genes encoding for the fourth component of the serum complement. Moreover, these units are located between a telomeric RP gene and a centromeric TNX gene, comprising the RCCX modules (RP-C4-CYP21-TNX) (Fig. 1)33). These genes are located in tandem and in an array (C4A, CYP21A1P, TNXA, C4B, CYP21A2, and TNXB). Genes C4A, C4B, CYP21A2, and TNXB all encode functional proteins, while CYP21A1P, TNXA, and RP2 genes are pseudogenes that do not encode proteins34).
The RCCX module shows high homology between the functional genes and the corresponding pseudogenes, leading to a wide variety of genetic rearrangement by unequal crossing over events, such as duplications, deletions, and fusions of the RCCX module 1,35).
To date, more than 200 CYP21A2 mutations have been discovered (http://www.hgmd.cf.ac.uk), and about 10 common mutations account for approximately 90% of cases4). More than 90% of CYP21A2 gene mutations are caused by gene conversion or unequal crossing over 36,37,38). Approximately 70%–75% of 21OHD cases are the result of the microconversion of the mutations in CYP21A1P to CYP21A21,39). About 20% are caused by unequal crossing over during meiosis, resulting in the deletion of a 30-kb gene segment38), encompassing the 3' end of the CYP21A1P, all of the adjacent C4B, and the 5' end of CYP21A2, producing the nonfunctioning chimeric CYP21A1P/CYP21A2 and chimeric TNXA/TNXB genes33). The remaining 1%–2% of affected alleles are spontaneous mutations not carried by either parent40).
Molecular analysis of the CYP21A2 gene
The genetic diagnosis of patients with 21OHD is not straightforward. However, it is a useful adjunct to hormonal measurements in the genetic counseling of parents upon the birth of a CAH child and of adolescents during the transition to adult care2). Therefore, molecular diagnosis of CYP21A2 mutations is recommended for genetic counseling and confirming the diagnosis in patients with the nonclassic form of 21OHD because neonatal screening may miss most cases of nonclassic CAH and adrenocorticotropic hormone stimulated 17OHP levels can be equivocal 2,6,41).
It should be stressed that molecular genetic diagnosis is more complicated for 21OHD than for many other monogenic disorders due to the high variability of the genomic region. This includes the coexistence of two or more mutations on the same allele or the presence of more than one CYP21/C4 repeat unit on the same chromosome. In addition, care should be taken to prevent genotyping the pseudogene because genetic results can be complicated due to the duplication, deletion, and recombination of CYP21A2 in the chromosome 6q21.3 region. Therefore, mutant alleles must be segregated in the parents to investigate their presence in different alleles and to verify de novo mutations4,42).
Several strategies have been developed for molecular analysis of CYP21A2, based on polymerase chain reaction (PCR)-driven amplification with allele-specific oligonucleotides to the CYP21A2 gene, followed by direct sequencing with assessment of the CYP21A2 gene copy number43). Sanger sequencing is the gold standard for detecting point mutations and small sequence variations (indels). However, large gene rearrangements cannot be detected by direct sequencing of PCR-amplified gene fragments33). The Southern blot method has traditionally been used to detect large gene deletions/conversions in the RCCX module. However, it is time-consuming, highly labor-intensive, and requires radioactive probes and a large amount of DNA. In addition, the Southern blot method has limitations in detecting chimeric RCCX modules, including CYP21A1P/CYP21A2 chimeric genes and TNXA/TNXB chimeric genes44).
Therefore, alternative methods have been developed. Recently, Multiplex ligation-dependent probe amplification (MLPA) analysis for the diagnosis of 21OHD has been increasingly used as an easy, simple, rapid, and sensitive tool to detect deletions or duplications of the CYP21A2 gene45). MLPA allows easy and rapid detection of gene copy number variations and the identification of chimerical genes in patients with 21OHD without using radioactive probes 45,46) and is thought to be a valid alternative to Southern blotting47). However, false positive results could occur because the mutations or polymorphisms very close to the probe binding regions and the ligation site may prevent probe hybridization and ligation47).
Prediction of clinical phenotype according to genotype
There is a wide spectrum of clinical features according to the type of mutations in CYP21A2. Depending on the residual activity of the mutation based on in vitro mutagenesis and expression studies, there is a good correlation between genotype and phenotype40). CYP21A2 mutations can be classified into 3 categories (A, B, C) according to the level of enzymatic activity predicted by in vitro transfection studies (Table 1)1,33,40). Group A consists of mutations such as deletions or nonsense mutations that totally ablate enzyme activity; these are most often associated with the severely affected SW form. Group B consists of the missense mutations, such as p.I172N, with 1%–2% of normal enzyme activity. These mutations are characteristically found in patients with the SV form. Group C includes mutations such as p.V281L and p.P30L that produce enzymes retaining 20%–60% of normal activity; these mutations are associated with mild, nonclassic CAH1,40).
Therefore, genotyping provides valuable diagnostic information by predicting of the clinical course of disease, and severe complications can be prevented, particularly in adults1,4,40). Approximately 65%–75% of CAH patients are compound heterozygotes with disease-causing mutations. Compound heterozygotes with two different CYP21A2 mutations usually have a phenotype compatible with the presence of the greater residual activity 40).
Conclusions
Early diagnosis of classic CAH is critical to save lives, and diagnosing nonclassic CAH is important to prevent unnecessary suffering. A baseline measurement of 17OHP levels can be used for screening and diagnosis of 21OHD. LC-MS/MS and GC-MS have recently been developed as highly sensitive and specific methods for targeted steroid hormone analysis. The detection of CYP21A2 mutations is important for clinical diagnosis because there is a high variability in 17OHP levels. Molecular genetic analysis of CYP21A2 is useful in confirming the diagnosis, providing genetic counseling, and predicting prognoses. Genotype is well correlated with the clinical severity of 21OHD. However, further research is needed to identify modifier genes in 21OHD, which could explain the phenotypic variability of androgen effects.