Introduction
Turner syndrome (TS) is one of the most common chromosomal disorders. It is caused by the loss of an X chromosome or the presence of a structurally abnormal X chromosome and has an estimated incidence of one in 2,500 girls born live1). The cardinal features, which occur in more than 90% of TS cases, are short stature and gonadal dysgenesis2). The patients with TS also have multisystemic complications such as congenital heart defects, skeletal anomalies, hearing impairment, ophthalmologic abnormalities, and renal structural anomalies2). In addition, TS patients manifest dysmorphic features such as high arched palate, micrognathia, low-set ears, epicanthal folds, lymphedema of hands and feet, and webbed neck2).
One candidate gene for short stature is located at the pseudoautosomal region 1 (PAR1) in the short arm of the X chromosome3). This gene was discovered on the distal part of PAR1, and was designated the short stature homeobox-containing (SHOX) gene or pseudoautosomal homeobox-containing osteogenic (PHOG) gene4,5). The SHOX gene is expressed in limbs, pharyngeal arches, osteogenic cells, and bone marrow fibroblasts, and is involved in skeletal growth and development6,7). The loss of the SHOX gene therefore leads to short stature and various skeletal abnormalities, such as short metacarpals, high-arched palate, cubitus valgus, Madelung deformity, and mesomelia6,7). The SHOX gene is expressed on both the inactive X chromosome and the active X or Y chromosome, thereby escaping from X chromosome inactivation4). According to the altered SHOX dosage, haploinsufficiency causes short stature, while overdosage contributes to tall stature.
Case report
The patient was born at term, without any perinatal problems, to nonconsanguineous Korean parents. Her prenatal evaluation was unremarkable. She did not experience any particular problem until her late teens.
At 17 years of age, she was brought to medical attention because of primary amenorrhea and the absence of secondary sexual characteristics. She was 183.5 cm (standard deviation score [SDS], 3.3) tall and weighed 77.4 kg (SDS, 2.0). Her body mass index (BMI) was 23 kg/m2 (SDS, 0.7). Her midparental height was 168.5 cm. Her serum luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels were 19.9 mIU/mL and 32.5 mIU/mL, respectively. Her serum estradiol was <10 pg/mL. Transrectal ultrasonography (US) showed a normal-sized uterus and adnexa. Her abdominal US and echocardiogram were normal. Hormone replacement therapy was initiated with 0.625 mg of conjugated estrogen on day 1-23 and 10 mg of medroxyprogesterone acetate on day10-23 every month. There was no menstruation after 6 months of medication, and the medications were replaced with 2.06 mg of estradiol hemihydrate for 14 days and the addition of dydrogesterone (10 mg) for the next 14 days every month. At 18 years of age, the follow-up transrectal US showed no specific findings. She manifested pubertal development (breast and pubic hair, Tanner stage 3). However, she still did not menstruate.
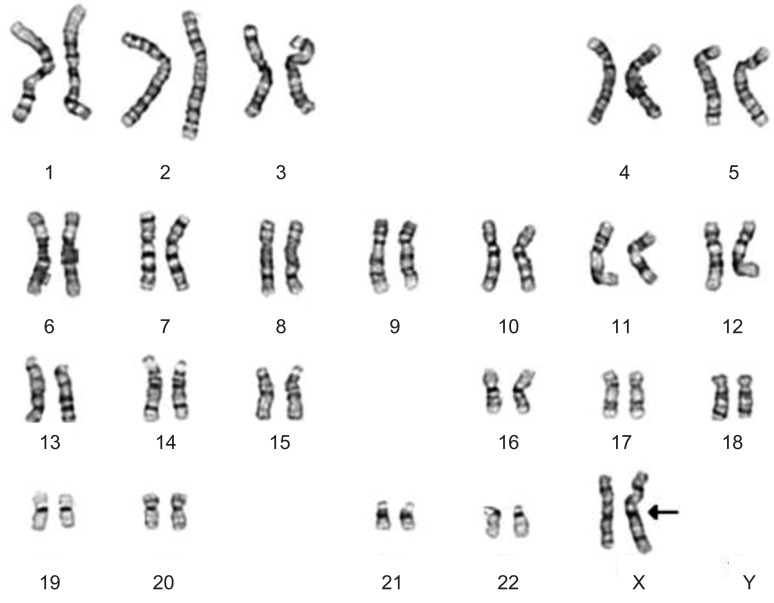
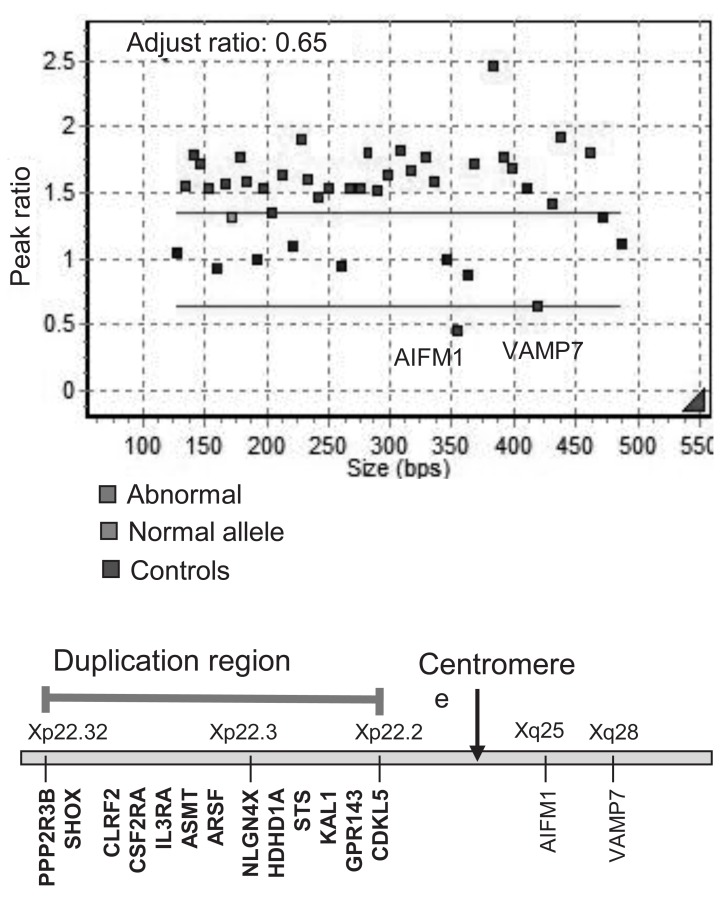
She was referred to a pediatric endocrinology clinic at the age of 29 years during hormone replacement therapy. She was 190 cm (SDS, 4.3) tall and weighed 101 kg (SDS, 3.3), with a BMI of 28.1 kg/m2 (SDS, 1.6). Her serum LH, FSH, and estradiol levels were 9.0 mIU/mL, 17.4 mIU/mL, and 18.1 pg/mL, respectively. Her serum triglyceride, total cholesterol, and high-density lipoprotein-cholesterol levels were 234 mg/dL, 138 mg/dL, and 34 mg/dL, respectively. Her blood pressure was normal. An x-ray of her left hand demonstrated complete epiphyseal fusion of the radius and ulna. Karyotype analysis revealed a nonmosaic pseudoisodicentric Xp chromosome, i.e., 46,X, psu idic(X)(q21.2) (Fig. 1). Multiplex ligation-dependent probe amplification (MLPA) analysis identified duplication of the Xp22.3-Xp22.2 region, encompassing the SHOX gene (Fig. 2).
She did not menstruate until she was 30 years old and still presented with breast and pubic hair at Tanner stage 3. Her weight had increased to 102.5 kg (SDS, 3.4) with a BMI of 28.4 kg/m2 (SDS, 1.7).
Discussion
This report describes the first Korean case of a TS patient who presented with tall stature due to SHOX overdosage. Approximately 45% of patients with TS have a 45, X karyotype, 10% have an isochromosome Xq, and the remaining patients have mosaicism or structural abnormalities of the X chromosome1). The present case had a nonmosaic 46, X, psu idic(Xq21.2) and presented with tall stature, surpassing the midparental height without other typical features of TS, except gonadal dysgenesis. The isochromosome X has a breakpoint at Xq21.2 and shows partial monosomy of Xq and partial trisomy of Xp. This implies a duplication of the terminal portion of Xp, including the SHOX gene. MLPA analysis confirmed the duplication from the PPP2R3 gene at the Xp22.3 to the FANCD gene at the Xp22.2 region (Fig. 2).
Several studies have demonstrated a correlation between SHOX overdosage and tall stature, as well as hypergonadotropic hypogonadism9,10). SHOX overdosage and gonadal dysgenesis contribute to sustained growth, with constant height velocity from infancy to adolescence9,10). The SHOX gene accelerates linear growth and inhibits fusion of the epiphyseal plate12,13), resulting in a marked increase in leg length and subsequent change in body proportion10). Although data are limited regarding the body ratio of the present case, a much longer lower than upper segment might be expected.
This patient continued to grow after 18 years of age, despite estrogen replacement therapy lasting 10 years, suggesting that SHOX overdosage surpasses the skeletal maturing effect of estrogen9). However, other deleted genes on Xq or duplicated genes on Xp could have contributed to the tall stature of our patient.
A TS patient with mosaicism that includes a 45, X cell lineage can show a normal height until the early teens and can be taller than the target height until the late teens. This finding suggests that cell lineages determining height may be 46,X,idic(X) or 46,X,der(X), rather than the 45,X cell lineage9,10,11). With this in mind, the present case showed tall stature until her late teens.
In summary, this report describes a TS patient with tall stature caused by a SHOX gene overdosage and estrogen deficiency, although tall stature could rarely be associated with TS in case of structurally abnormal X chromosome containing an extra Xp.