Introduction
Gigantism refers to excessive secretion of growth hormones (GH) that occurs during childhood when epiphyseal growth plates allow for excessive linear growth. On the other hand, acromegaly is the same phenomenon as gigantism but occurring in adulthood1). These two disorders may partially overlap depending on the developmental stage. Approximately 10% of acromegalics are shown to exhibit tall stature and the majority of giants eventually demonstrate the features of acromegaly2). The mean age for the onset of acromegaly is within the 3rd decade of life, whereas gigantism may begin at any age prior to epiphyseal fusion1). True gigantism is extremely rare, and it is usually caused by a pituitary adenoma3,4,5).
Pituitary adenomas occur with an annual incidence of 20 cases per million, with adenomas derived from somatotrophs and secreting GH accounting for 3 cases per million6). GH-secreting adenomas seem to be more invasive and aggressive in children than in adults5,7). Surgery has traditionally been the first line of treatment, with radiation reserved for inoperable cases. Also, medical therapy has taken on an important role in the management of patients with GH excess with development of somatostatin analogues8). We present two cases of gigantism caused by a GH-secreting pituitary adenoma with clinical and microscopic findings.
Case reports
Case 1
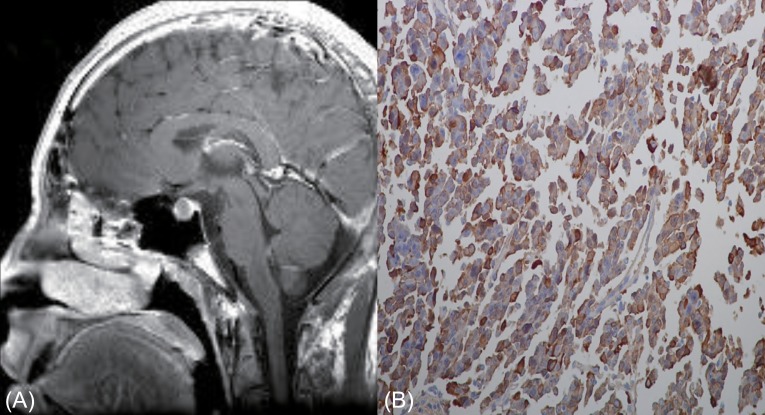
A 14.7-year-old boy was presented to Chonnam National University Hospital because of extremely tall stature. His height was 192.0 cm (14 cm above the 97th percentile) and body weight was 70.5 kg (90th-97th percentile). He showed enlarged hands and feet, and prognathic mandibles. His body proportion was normal and his pubertal stage was mature (Tanner stage 5). Bone age was normal for chronological age according to the method of Greulich and Pyle9). There was no family history of tall stature (father, 176.0 cm; mother, 167.0 cm) or any endocrine diseases. Laboratory investigation showed the following results; random serum GH, 38.4 ng/mL (normal range, 0-5 ng/mL); insulin-like growth factor 1 (IGF-1), 624.0 ng/mL (normal range for age, 220-616 ng/mL); IGF-BP-3, 6,301.6 ng/mL (normal range for age, 2,200-5,900 ng/mL); and prolactin, 8.94 ng/mL (normal range, 3-18 ng/mL). GH failed to suppress during an oral glucose tolerance test (OGTT; nadir serum GH, 22.7 ng/mL [normal range, <1 ng/mL]). Magnetic resonance imaging (MRI) of the brain revealed a 12-mm-sized pituitary adenoma (Fig. 1A). Neither pituitary insufficiency nor visual impairment was present. Transsphenoidal surgery was perfor-med and a pathologist reported a pituitary adenoma with positive immunohistochemical staining for GH (Fig. 1B). A pituitary MRI scan performed 4 months after surgery showed recurrence/residual tumor with a size of 5 mm and a basal GH level of 7.1 ng/mL (normal range, 0-5 ng/mL). Medical treatment with intramuscular injection of the long-acting somatostatin analogue octreotide LAR (Sandostatin LAR, Novartis Pharma AG, Basle, Switzerland) at a dose of 20 mg was given every 4 weeks. Six months after medical treatment, the serum GH levels increased further, and nadir GH during OGTT was 6.6 ng/mL (normal range, <1 ng/mL). Therefore, the patient underwent secondary surgery. Three months after reoperation, the GH level was 0.2 ng/mL and IGF-1 was 205 ng/mL (normal range, 220-616 ng/mL). During 2 years' follow-up, adrenal, gonadal and thyroid functions remained unchanged and annual MRI showed no signs of relapse. GH and IGF-1 levels also showed within normal range. His height was 200.8 cm (>97th percentile) at age of 16.7 years.
Case 2
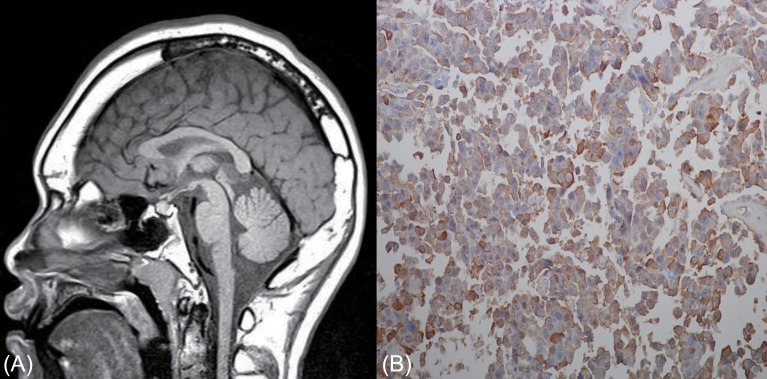
A 14.9-year-old boy was referred to our pediatric clinic due to extremely tall stature. His height was 197.3 cm (19 cm above the 97th percentile) and body weight was 101.5 kg (20 kg above the 97th percentile). He showed enlarged hands and feet, and prognathic mandibles. His body proportion was normal. The pubertal status was mature (Tanner stage 5). He showed advanced bone age of 16.0 years according to the method of Greulich and Pyle9). There was no family history of tall stature (father, 175.0 cm; mother, 163.0 cm) or any endocrine diseases. Endocrine investigation revealed the following: basal serum GH, 9.3 ng/mL (normal range, 0-5 ng/mL); IGF-1, 518.0 ng/mL (normal range for age, 220-616 ng/mL); IGF-BP-3, 3,450.0 ng/mL (normal range for age, 2,200-5,900 ng/mL); and prolactin, 10.39 ng/mL (normal range, 3-18 ng/mL). GH failed to suppress during an OGTT (nadir serum GH, 9.0 ng/mL [normal range, <1 ng/mL]). A pituitary MRI scan showed a 6-mm-sized pituitary adenoma (Fig. 2A). Transsphenoidal surgery was performed and histopathological examination demonstrated a pituitary adenoma with positive immunohistochemical staining for GH (Fig. 2B). Three months after surgery, the GH level was 0.2 ng/mL (normal range, 0-5 ng/mL) and nadir GH during OGTT was less than 0.1 ng/mL (normal range, <1 ng/mL). Pituitary MRI scans at 6 months postsurgery showed no residual tumor. During 4 years' follow-up, adrenal, gonadal and thyroid functions remained unchanged and annual MRI showed no signs of relapse. GH and IGF-1 levels also showed within normal range. His final height was 206.0 cm (>97th percentile) at age of 19 years.
Discussion
Hypersecretion of GH in childhood causes gigantism with potential clinical symptoms including accelerated growth velocity with tall stature, enlargement of the hands and feet, coarsening of facial features, and headaches. Most cases are caused by benign pituitary adenomas3,4,5). Pituitary gigantism is very rare and the description of the disease is limited to small series and case reports. Approximately 100 cases of children with pituitary gigantism have been reported5,10,11). The median age at diagnosis is 12 years, even though the median age of initial signs and symptoms is 8 years. Even a congenital onset of GH excess has been suggested by linear growth acceleration occurring within the first month of life in children with documented gigantism11). There are a few cases of pituitary gigantism have been reported in Korean children and adolescents12,13). Our patients were 14 years of age at the time of diagnosis and presented with extremely tall stature.
Biochemical features of children with gigantism are similar to those of acromegalic adults including elevations in serum GH and IGF-1 levels, and failure to suppress GH level after OGTT. GH level is reflective of neurosecretory dysfunction which is characteristic of GH-cell adenomas14), while IGF-1 level provides a surrogate marker of peripheral GH bioactivity15). The gold standard for making the diagnosis of GH excess is failure to suppress serum GH level to less than 1 ng/mL after OGTT1). Hyperprolactinemia is a common finding in GH excess presenting in childhood, undoubtedly related to the fact that mammosomatotrophs (GH and prolactin-secreting cells) are by far the most common type of GH secreting cells involved in childhood gigantism1). However, gigantism caused by a pituitary tumor comprised of somatotropes (GH-secreting cells) show a normal prolactin level1). Pituitary imaging by MRI or computed tomography is an essential step in the evaluation following biochemical detection of GH excess.
Several therapeutic modalities have been used for the treatment of GH excess. For well-circumscribed pituitary adenomas, transsphenoidal surgery is the recommended treatment and it may be curative3,5,7). Transshpenoidal pituitary surgery has been shown to be safe in both pediatric patients with gigantism and in adults with acromegaly5). On the other hand, putative pituitary surgery damage with lifelong hormonal replacement therapy has to be taken into account16).
Radiation therapy, used as adjunctive or primary treatment, has also been moderately successful in inducing normalization of GH level. However, the efficacy of radiation therapy in decreasing GH secretion in acromegalic patients is delayed, with a reduction of approximately 50% by 2 years and 75% by 5 years17). Another major concern of irradiation is a high incidence of hypopituitarism after therapy1).
The somatotropin analogue, octreotide, has been found to be effective in the treatment of acromeglaic patients with GH excess. A sustained-release somatostatin analogue has also been shown to be successful in returning GH levels to normal in acromegalic adults with pituitary adenomas18). Octreotide suppressed GH secretion and normalized IGF-1 levels in 50%-70% of patients, and reduced the size of the tumor in most patients19). Good results of octreotide therapy have been reported in children11,16). In our patient (case 1), the tumor did not respond well to this treatment. Therefore, he underwent transsphenoidal reoperation. Recently, treatment of acromegaly with a new GH receptor antagonist pegvisomant has been introduced. Pegvisomant is a GH analogue that binds to GH receptors on the cell surface, and blocks GH receptor dimerisation8). Pegvisomant has been effective in acromeglaic patients resistant to somatostatin analogues8). After further studies, pegvisomant might be an additional option for the treatment of pituitary gigantism in children.
In conclusion, we report here two cases of childhood gigantism caused by GH secreting pituitary adenomas. Treatment of pituitary gigantism in childhood is difficult and often unsatisfactory. Our patients should be closely followed up for the potential risk of hypopituitarism.