Introduction
Turner syndrome (TS) is the most common chromosomal aneuploidy that affects 1 in every 2,000 girls, and is characterized by short stature and gonadal dysgenesis in females who lack all or part of one X chromosome1). Approximately 50% of patients with TS have complete loss of one X chromosome, whereas the rest of patients with TS display mosaicism or structural abnormalities of the X chromosome, for example, 46, X, i(Xq), 46, X, del(X), 46, X, r(X), etc.2). Like complete monosomy X, partial deletions of either the short arm or long arm can cause features of TS. Many studies have been conducted to verify and delineate the proposed loci for genes pertaining to the TS phenotype. Some studies have indicated that the genes for physical and cognitive features lie on Xp, whereas genes for ovarian function are present on both Xp and Xq3,4,5). The correlation of thyroid autoimmunity with the type of karyotype abnormality and loci of gene is not well-described with conflicting results6,7,8,9). Approximately one third of girls with TS may enter spontaneous puberty, but only half those completed with menarche10). The prevalence of spontaneous puberty is higher in patients with mosaic TS. And there were a few rare cases10,11,12,13,14) about TS with precocious puberty.
The authors report a 17-year-old TS variant patient with early normal puberty and Graves disease, who had no clinical features of TS during puberty.
Case report
A 17-year-old girl visited the hospital with the chief compliant of goiter with acute onset. She was born via spontaneous vaginal delivery at 37-week gestation, weighing 2.6 kg. She was the second child of unrelated healthy parents. Her father was 171 cm tall and her mother was 158 cm tall. Midparental height was 158 cm.
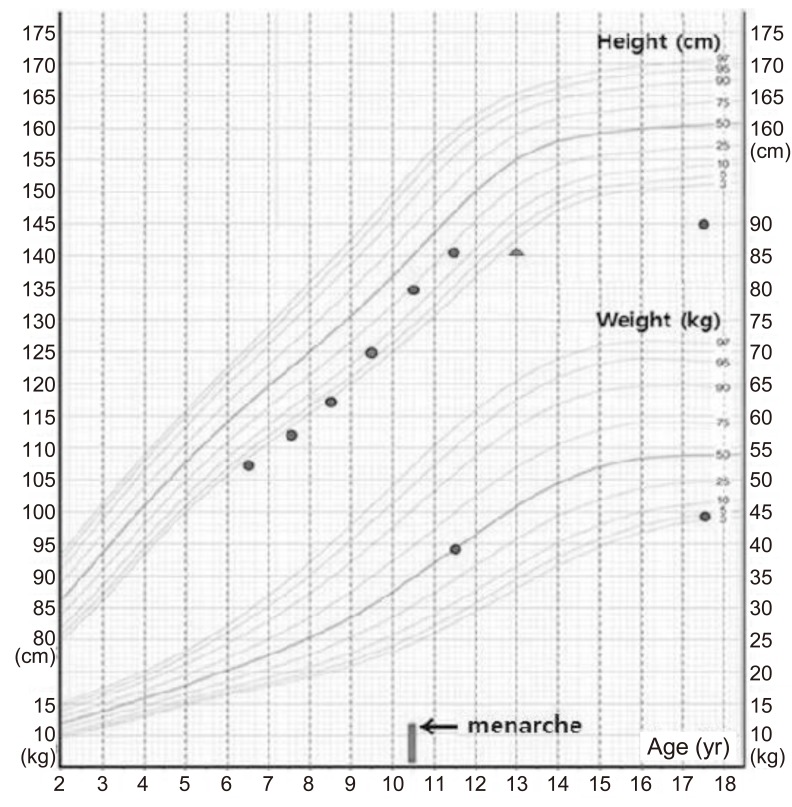
She had visited our clinic for growth evaluation at 11 years 10 months of age, when her height was 141.2 cm (10th-25th percentile) and her weight was 39.7 kg (50th percentile). Tanner stage was breast 4 and pubic hair 4. Her bone age was approximately 13 years and her height for bone age was <3rd percentile. Menstruation had been started at 10 years 9 months of age. According to her school record, she was short before pubertal onset, but her height and weight were relatively normal during puberty (Fig. 1). So, idiopathic short stature and early normal puberty were diagnosed at 11 years 10 months of age.
At the age of 17 years, goiter developed abruptly, and the patient visited the hospital again. She was short in stature and of normal weight, measuring 144.7 cm (<3rd percentile) and weighing 44 kg (3rd-5th percentile). She showed no signs of TS, such as webbed neck, short neck, cubitus valgus, high arched palate, and shield chest deformity, except for short stature. Her menstrual cycle was regular. Tanner stage was breast 5 and pubic hair 5. She had normal intelligence and high grades in school.
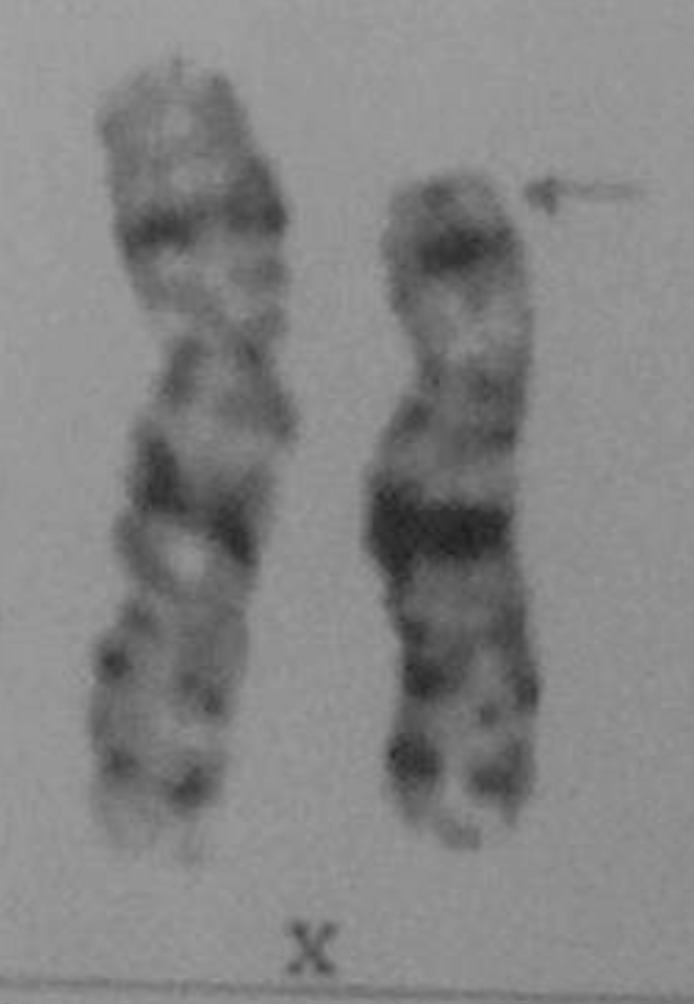
The laboratory profiles demonstrated normal complete blood count, chemistry panel, and electrolytes. The thyroid function test showed: thyroid stimulating hormone (TSH), 0.01 mIU/L (reference, 0.25-4.0 mIU/L); free thyroxine, 7.3 ng/dL (reference, 0.7-2.0 ng/dL); triiodothyronine, 680 ng/dL (reference, 60-190 ng/dL), thyroglobuin antibody, 7.06 U/mL (reference, 0-0.3 U/mL); thyroid stimulating immunoglobulin, 22.7% (reference, 0%-15%); microsomal antibody, 1.45 U/mL (reference, 0-0.3 U/mL), suggesting Graves disease. On thyroid ultrasonography, enlargement of both thyroid glands with increased vascularity was observed. A Technetium-99m thyroid scan showed diffuse, enlarged thyroid glands with markedly increased uptake. Other results were as follows: luteinizing hormone (LH), 14.65 mIU/mL (reference: follicular phase, 0.6-6.2 mIU/mL; luteal phase, 0-6.0 mIU/mL); follicle stimulating hormone (FSH), 7.78 mIU/mL (reference: follicular phase, 3.3-8.8 mIU/mL; luteal phase, 1.6-8.7 mIU/mL); and estradiol, 156.99 pg/mL (reference: follicular phase, 60-200 pg/mL; luteal phase, 60-260 pg/mL), without evidence of ovarian failure. Because the final height was below the 3rd percentile with coexisting thyroid dysfunction and autoimmunity, chromosome analysis was performed. The karyotype was 46,X,del(X)(p22.1), one of the form of TS (Fig. 2). Methimazole was prescribed for the patient and a regular thyroid function test was performed.
Discussion
Loss of interstitial or terminal long-arm material of the X chromosome (Xq) can result in short stature and primary or secondary ovarian failure15). However, absence of the long arm of the X chromosome with normal stature suggests that the presence of the short arm of the X chromosome maintains the stature of the affected patient1). Deletion involving the long arm of the X chromosome generally results in ovarian failure if the proposed critical region Xq13-q26 is involved1). Deletion of the whole short arm of the X chromosome (Xp) in females is associated with short stature, ovarian failure, and the classic stigmata of TS. The prevalence of an Xp deletion among patients with TS is 2%16). Patients with terminal Xp deletion have short stature and may have some somatic traits of TS; gonadal function is generally preserved16). Women with more distal deletions [del(X)(p21.1 to p22.1 or p22.2] menstruate more often, but many are infertile or have secondary amenorrhea17). Thus, Xp [X(pter->p21] clearly plays a role in ovarian development. Therefore, we should perform follow-up regarding fertility or occurrence of secondary amenorrhea even in a patient with distal Xp deletion, although our patient had normal puberty and menstruation in adolescence. The Xp22.3-Xp22.12 region is located at the Xp terminus and includes the SHOX gene. The SHOX gene is located at the very tip of the short arm of both sex chromosomes, inside the telomeric portion of pseudoautosomal region 1, containing genes that escape X inactivation. The X-linked zinc finger protein (Zfx) is also located at Xp22.1-21.3, and is a candidate gene for short stature and ovarian failure17). Most TS patients with Xp deletion have short stature, considering statural determinant17). However, this patient's height was in the normal range (10th-25th percentile) from 9 years to 12 years and she experienced short stature after puberty. The exact cause of normal height during the period is unclear, and maybe because of the effect of intact Xq chromosome and early normal puberty, respectively. There are only six cases10,11,12,13,14) about central precocious puberty in patient with TS have been reported. Five of them showing mosaic TS and one of them showing structural abnormality of one X chromosome like our patient. The mechanism is unclear, and may be the result of abnormalities in the hypothalamic feedback system with increased levels of gonadotropins, FSH surge before ovarian failure, or elevated levels of TSH by interaction between TSH and the human FSH receptor. All had normal height according to standard growth curve when diagnosed as TS, like this case. When precocious puberty is combined with TS or if midparental height is tall, the patient may have normal height and the diagnosis may be delayed12). More studies are needed regarding the correlation between TS and central precocious puberty. With regard to thyroid autoimmunity, correlation with karyotype and phenotype shows variable results7). In some of the studies, especially in girls with isochromosome Xq, levels of thyroid autoantibody are the highest7,8), but in others9), the results did not reach statistical significance.
In conclusion, we experienced a case of TS with Xp22.1 deletion presenting with short final stature, early normal puberty, and Graves disease, without short stature during puberty. We suggest that even though a patient's puberty is normal, when the final height is short and autoimmune thyroid disease is accompanied, chromosome analysis for TS should be considered. Further research about determinant factors and genes on the X chromosome in growth, ovarian function, and autoimmunity is needed. Additionally, screening karyotypically normal individuals with TS features for mutations in candidate genes is necessary.