Introduction
Congenital central hypothyroidism (C-CH) is an unusual condition characterized by low levels of both thyroid hormones and of thyroid-stimulating hormone (TSH). Patients with this disorder cannot be identified by neonatal screening programs based on TSH measurements1,2). However, neonatal screening for CH on the basis of thyroxine (T4) or free T4 and TSH concentrations can be performed to diagnose this type of CH1,2,3,4,5,6).
C-CH is caused by mutations of transcription factors involved in pituitary development and differentiation, including POU1F, PROP1, HESX1, LHX3, and LHX47,8,9). Because more than one pituitary cell type is deficient in these genetic diseases, patients with mutations of transcription factors often present with as a multiple pituitary hormone deficiency7,8,9). On the other hand, C-CH presenting in the absence of other syndromes remains a rare disease and is due mainly to a genetic deficit of the β-subunit of TSH (OMIM 188540)10,11) or mutations of the thyrotropin-releasing hormone (TRH) receptor (OMIM 188545)12,13). Recently, it has been demonstrated that deficiency of immunoglobulin superfamily member 1 (IGSF1) (OMIM 300888) can also be a cause of C-CH14,15,16,17). This review will focus on clinical findings in patients with mutations of IGSF1, and the molecular basis of their disease.
Immunoglobulin superfamily member 1
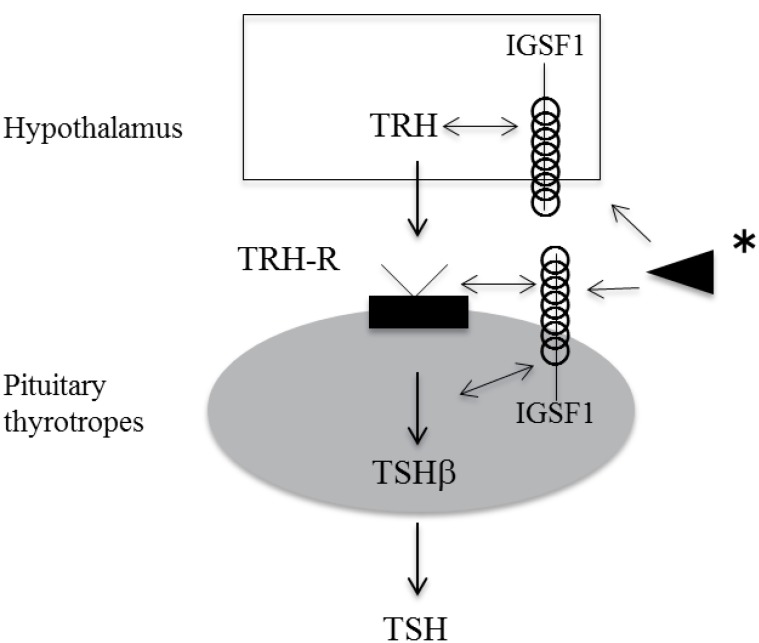
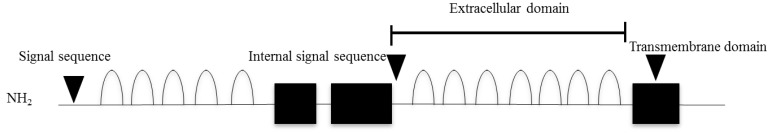
IGSF1 is a plasma membrane immunoglobulin superfamily glycoprotein18,19). This protein has a putative signal peptide and twelve C2 type immunoglobulin (Ig)-like domain loops, a transmembrane domain, and a short intracellular C tail19) (Fig. 1). The protein is cotranslationally cleaved such that only the C-terminal domain, containing 7 Ig loops, reaches the plasma membrane19). The gene encoding IGSF1 is located on Xq 26.218). Human IGSF1 and murine Igsf1 mRNAs are highly expressed in Rathke's pouch and in adult pituitary gland and testis14). Moreover, IGSF1 protein is expressed in murine thyrotropes, somatotropes, and lactotropes, but not in gonadotropes or in the testis14).
IGSF1 was initially hypothesized to be a candidate for the inhibin coreceptor in the pituitary gland, and it was therefore designated inhibin binding protein or p120.520). However, Igsf1 knockout mice showed no alternation of follicle stimulating hormone synthesis or secretion, and normal fertility21). Moreover, a recent in vitro study did not demonstrate binding of inhibin and IGSF122). Thus, the physiological function of IGSF1 remains unknown.
Members of the IgSF have a wide variety of functions, acting as cell surface antigen receptors, coreceptors and costimulatory molecules of the immune system, molecules involved in antigen presentation to lymphocytes, cell adhesion molecules, certain cytokine receptors, and intracellular muscle proteins23,24,25,26,27,28). Some IgSF members play crucial roles not only in nervous system development but also in the adult during neural repair and synaptic plasticity26,27,28). Considering the function of other IgSF family members, IGSF1 may also act as a signal transduction molecule and/or cell adhesion molecule in the pituitary.
Neonatal screening for C-CH in Japan
In Sapporo city, the neonatal screening program for congenital hypothyroidism has employed the measurement of free T4 and TSH in the same filter-paper blood spot since 1986. This system has enabled us to identify C-CH in the neonatal period. Between January 2000 and December 2004, 83,232 newborns were screened and six C-CH patients identified as a result of follow-up of low free T4 and nonelevated TSH screening test results (a frequency of one in 13,872)4). Clinical characteristics and neonatal screening data of these patients are summarized in Table 1. Four showed multiple pituitary hormone deficiencies with pituitary malformations on magnetic resonance imaging. One patient was diagnosed as having Prader-Willie syndrome. Of these 6, the C-CH of a single one was caused by IGSF1 deficiency.
Adachi et al.6) have recently reported the prevalence of C-CH in Kanagawa prefecture between 1999 and 2008. According to their study, neonatal screening for free thyroxine and TSH identified 24 C-CH patients, 14 of whom had multiple pituitary hormone deficiencies, eight had isolated C-CH, and two had undetermined pituitary involvement. Thus, the prevalence of CH-C was estimated at 1 in 30, 833 live births. During this period, 213 patients with CH of thyroid origin were diagnosed. Thus, C-CH constituted 10% of permanent CH detected by neonatal screening.
Lanting et al.3) have reported the results of a neonatal screening program based on initial T4 and subsequent TSH determination with a T4 binding globulin in the Netherlands. In their study, among 385,000 infants screened during the period 1995-2000, the incidence of C-CH was 1:16,404 with 75% of C-CH diagnosed as having multiple pituitary hormone deficiency. This suggests that the incidence of isolated C-CH is likely to be around 1:65,000. Moreover, Joustra et al.17) reported an IGSF1 defect in 8 of 11patients with isolated C-CH. Therefore, it is postulated that the incidence of C-CH caused by IGSF1 deficiency is approximately 1:100,000.
As mentioned above, in our small study over the short period 2004 to 2008, only one patient had a IGSF1 deficiency4). Thus, the incidence of IGSF1 deficiency was 1 in 80,000, but the exact prevalence of C-CH caused by IGSF1 deficiency in Japan cannot be said to have been determined. Further study is required.
Phenotypes associated with IGSF1 deficiency
In a previous report, six familial cases of C-CH in the Netherlands and one in Italy were identified by neonatal screening14). We have identified three IGSF1-deficient patients by neonatal screening15,16). 123I scintigraphy showed a normalsized thyroid in our patients. The Dutch study also showed normal-sized thyroids by ultrasound, except in one case14).
In our study, one patient who was not identified by TSH screening in the neonatal period suffered growth retardation, and here, medical examination led to the diagnosis of growth hormone (GH) deficiency and central hypothyroidism at 4 years of age. We also diagnosed GH deficiency in one other patient. Joustra et al.17) reported 3 patients with GH deficiency together with CH, but when GH secretion was reevaluated in two patients at adolescence, it was shown to be normal. It is possible that untreated hypothyroidism may impair GH secretion. Alternatively, because IGSF1 is expressed in somatotropes14), an IGSF1 defect in these cells may affect GH secretion.
Four of our 5 patients had low levels of prolactin. The Dutch study found that 16/24 had hypoprolactinemia17). Thus, prolactin deficiency is one of the clinical features of IGSF1 deficiency.
Because TSH-based neonatal screening cannot identify patients with IGSF1 deficiency, treatment with thyroxine is delayed in those patients undergoing TSH screening. Therefore, there is a concern that psychomotor development may be delayed by late onset of treatment. However, several family members of affected patients showed no symptoms and their hypothyroxinemia was mild14,17). Their intelligence quotient seemed to be within the normal range. By contrast, in our study, one patient whose treatment was started at 4 years of age showed slightly delayed development16). Thus, it is still possible that severe cases of IGSF1 deficiency might be accompanied by developmental delay. Three of our patients did have delayed puberty and their serum luteinizing hormone and FSH levels remained at prepubertal levels even at 12 years of age. In the report of Sun et al.14), 10 of 11 evaluated patients showed delayed testosterone production and pubertal growth. Delayed secondary sex characteristics may therefore be one of the clinical hallmarks in patients with pathogenic IGSF1 mutations. In addition, testicular enlargement after adolescence is a characteristic feature of IGSF1 deficiency. It has been reported that all such patients aged more than 12 years manifested macroorchidism. In our study, two patients showed increased testicular size at 13 years of age (14 mL according to the Prader orchidometer) but one other had normal sized testes at 17 years of age. It has been suggested that testicular size increases from young to late adulthood. Thus, in this case testicular size may be increased in the future. Despite macroorchidism, fertility of male patients is likely to be normal14,17).
We performed a TRH test twice in one patient. Compared with the result of the first TRH test, serum TSH was markedly reduced in the second test16). It has been observed that in Igsf1 knockout mice, pituitary TSH synthesis and secretion are reduced, and pituitary TRH receptor mRNA expression is decreased14). It was speculated, therefore, that reduced TRH receptor signaling might be one of the pathogenic mechanisms of C-CH in IGSF1-deficient patients. TRH signaling through the TRH receptor is known to be an important factor for normal proliferation of thyrotropes29,30), so in patients with IGSF1 defects, their proliferation during postnatal development may be impaired. This may indicate that IGSF1 is involved in thyrotrope proliferation and differentiation, in addition to TSH secretion (Fig. 1).
Joustra et al.17) have analyzed metabolic parameters of children and adolescents with IGSF1 deficiency. They found that body mass index (BMI) was increased in 3 of 7 patients, and percentage of fat was high in 5. In adult cases, most patients showed increased BMI and fat, but this is not likely to be due to hypothyroidism, because patients who were treated from early infancy also showed these findings, as well as untreated individuals. This may suggest that obesity is not caused by hypothyroidism but rather a direct effect of IGSF1 deficiency itself. In our studies, the BMI of three patients was 18.5, 24.0, and 25.2 kg/m2 at 12 years of age. Two patients showed higher BMI for their age than normal Japanese children31), and thus careful follow-up for metabolic parameters is also necessary in IGSF1 deficiency.
Mutations/deletions of IGSF1
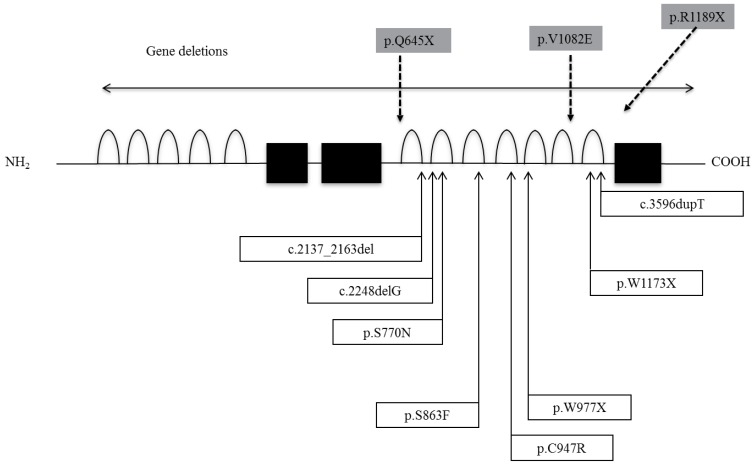
Sun et al.14) identified 8 distinct mutations and 2 deletions in IGSF1 in 11 families (Fig. 2). The mutations included in-frame deletions, single nucleotide deletions, nonsense mutations, missense mutations and one base duplication. In vitro expression studies of several mutations to analyze the functional consequences demonstrated that the encoded proteins migrated predominantly as immature glycoforms and were largely retained in the endoplasmic reticulum, resulting in decreased membrane expression14). In Japanese patients, we have reported two nonsense mutations, one missense, and one base insertion mutation (Fig. 3). In addition, a large deletion and a splice junction mutation have been identified (unpublished data). Two patients had the R1189X mutation. From interview, they were not related, but they lived in the same prefecture and thus this mutation may be a founder effect. We also performed an in vitro study and found that the V1082E mutated product was severely impaired in its posttranslational modification and membrane trafficking, similar to results of S770N, S863F and C947R, which were previously reported.
Conclusions
IGSF1 deficiency is a newly-discovered cause of C-CH. The physiological role of IGSF1 is unknown, and when clarified in future, its role in the mechanisms responsible for a variety of symptoms such as hypothyroidism, PRL deficiency, macroorchidism and delayed puberty will be clearer. IGSF1 is important for the pituitary-thyroid axis and the development puberty and thus represents a new player controlling growth and puberty in childhood and adolescence.