Introduction
Denys-Drash syndrome (DDS, ONIM number 194080) is a complex disorder characterized by nephropathy, Wilms tumor and 46,XY disorders of sex development (DSD)1,2). The abnormalities identified in DDS have been attributed to mutations in the WT1 gene3,4,5,6). The finding of 46,XY DSD, during the neonatal period, is suggestive of the diagnosis of DDS. Anti-Mullerian hormone (AMH), also known as Mullerian inhibiting substance (MIS), a member of the transforming growth factor of the transforming growth factor-╬▓ superfamily produced by Sertoli cells of the fetal testis from 7 weeks gestation, is responsible for the regression of Mullerian ducts in male fetuses, the first step of male sex differentiation of the genital tract.
Because Wilms tumor 1 (WT1) gene product suppress AMH gene, mutations in WT1 can cause persistence of the Mullerian duct in men7). AMH begins to be expressed in a sexually dimorphic pattern during embryo development. In mice, AMH is expressed in male embryonic Sertoli cells from embryonic day 13 until birth8).
Here, we report a neonate with double uterus of DDS and a low level AMH firstly checked in human patient with DDS.
Case report
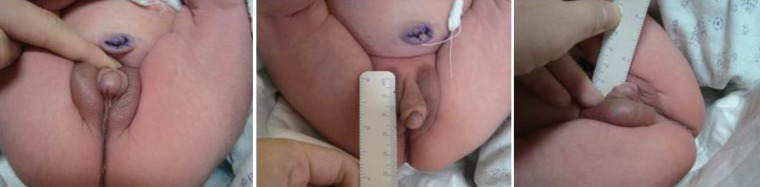
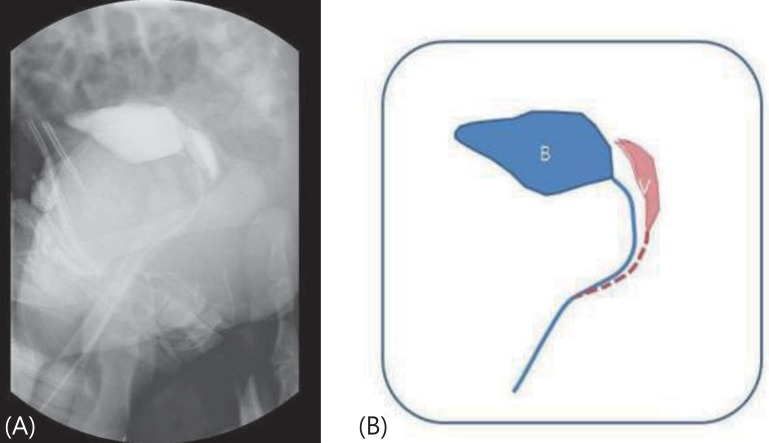
A 2-day-old Korean newborn was referred to Gyeongsang National University Hospital with ambiguous genitalia (hypospadias without testis). The baby was born after 39 weeks of gestation and weighed 3,000 g. There were no perinatal problems. The external genitalia length of patient was 2.5 cm sized and urethral opening was placed at ventral side. Testis was not palpable with pigmented scrotum like structure (Fig. 1). On admission to our hospital, the blood pressure was 86/48 mmHg. The initial laboratory tests revealed; hemoglobin, 16.7 g/dL, blood urea nitrogen, 15 mg/dL, serum creatinine, 0.2 mg/dL, albumin, 3.4 g/dL, total cholesterol, 133 mg/dL, and sodium, 146 mEq/L. The urinalysis was normal. The echocardiogram showed no cardiac abnormality. The abdominal ultrasound and voiding cystography showed seminal ducts, bilateral testes in the inguinal areas and a double uterus located between the bladder and the rectum (Fig. 2). The karyotype was 46, XY. We analyzed WT1 the mutation in this patient after obtaining informed consent from the parents. The WT1 gene was evaluated after the extraction of genomic DNA from peripheral blood nucleated cells using a commercial kit (QIAamp DNA Blood Mini Kit, QIAGEN, Hilden, Germany). Exons 1, 47 and 10, with their bilateral flanking introns, were amplified individually from the genomic DNA by polymerase chain reaction (PCR). Exons 1-10, with their bilateral flanking introns, were amplified individually from the genomic DNA by PCR. The PCR products were sequenced directly. A heterozygous missense mutation, p.D464N (=p. Asp464Asn), reported as p.Asp396Asn, was detected in exon 8 of the WT1 gene (Fig. 3).
The AMH level was 6.24 ng/mL (normal range, 375-625 ng/mL)9).
After the diagnosis of DDS the patient was discharged and followed-up as an outpatient with conservative management. The renal function, growth and development remained normal at the last follow-up at 10 months of age.
Discussion
A recent study reported that the WT1 gene controls MIS expression by inhibiting AMH secretion. The WT1, SRY box protein 9, and steroidogenic factor 1 could synergistically activate the MIS promoter, but without WT1, MIS promoter was not activated. The WT1 is an essential factor for activation of the MIS promoter; therefore, the persistence of the Mullerian duct in patients with DDS may result from deregulation of the MIS gene10).
This is the first case study to measure the AMH levels in a patient with DDS. DDS is characterized by a wide variety of genital anomalies including 46,XY DSD. The external genitalia of patients can be female, male or ambiguous. In addition, the internal genitalia can vary and can include normal-sized testes in the normal position together with fully feminized genital organs (vagina, cervix, and uterus); however, there can also be abnormal-sized testes in abnormal positions together with various combinations of duplicated internal genital organs. The persistent Mullerian duct syndrome is in the differential diagnosis of ambiguous genitalia; these patients have male external genitalia, but do not have duplicated genitalia as in DDS.
It is unclear why duplicated genitalia are found in the DDS but not in the persistent Mullerian duct syndrome; both syndromes have been associated with low level of AMH. Previous study showed that the level of AMH in the persistent Mullerian duct syndrome is 0-10 pg/mL, so low level of AMH can not discriminate these two clinical condition.
The mutation identified in this patient had been previously described with DDS11,12,13,14). However, this is the first case of DDS with a bicornate uterus (Table 1). DDS with duplicated internal genital organs has been reported with several different mutations15). However, duplicated internal genitals are not always present in patients with DDS.
Although many different WTI mutations have been identified with DDS, there is no clear genotype-phenotype correlation to date15). However, our case suggests that measurements of AMH in patients with DDS might improve our understanding of the association between WT1 and the MIS gene and their role in genital development.