Introduction
Congenital lipoid adrenal hyperplasia (CLAH) is the most severe form of congenital adrenal hyperplasia that is caused by mutations in the steroidogenic acute regulatory protein (StAR)1,2). The mutations in StAR gene resulted in failure of the transport cholesterol into mitochondria for steroidogenesis in the adrenal gland and gonad. Patients with CLAH usually manifest clinical symptoms including hyponatremia, hyperkalemia, hypoglycemia, acidosis, cholestasis, enlargement of adrenal gland, feminization of external genitalia and severe salt wasting such as dehydration and vomiting.
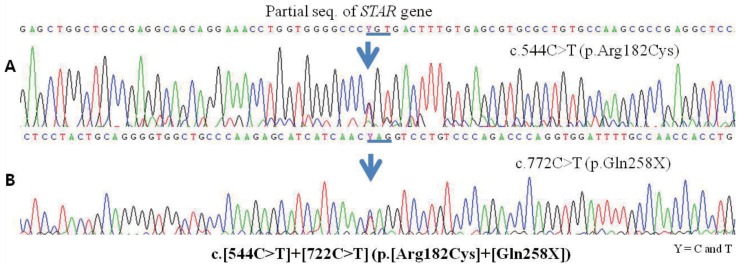
We present a case of CLAH that showed different cortisol levels and manifestations in twins. Patient A was admitted with adrenal crisis symptoms at 38 days of age. Although patient B showed only hyponatremia and hyperpigmentation without adrenal insufficiency symptoms, laboratory results showed lower level of cortisol and higher level of adrenocorticotropin hormone (ACTH). Gene analysis was conducted because of adrenal insufficiency symptoms combined with normal level of 17-hydroxyprogesterone (17-OHP), low level of cortisol, and increased ACTH level which was suggestive of CLAH. The results of gene analysis of StAR in the twin revealed the same heterozygous conditions for c.544C>T (Arg182Cys) in exon 5 and c.722C>T (Gln258*) in exon 7. It was the first case of mutation of p.R182C in exon 5 of the StAR gene in Korea.
Case report
Phenotypic female twins (A, B) were born at 36+2 gestational week to unrelated parents. Apgar scores were 8 at 1 minute and 9 at 5 minutes in both patients. Birth weight was 1,920 g (<10 percentile) in A and 2,040 g (10 to 25 percentile) in B, height was 43 cm (10 to 25 percentile) in A and 44 cm (10 to 25 percentile) in B. Patient A needed the oxygen supplementation with hood and nasal prong for 3 days due to desaturation and tachypnea.
They had symptoms as hyperpigmentation, slightly elevated potassium level and low level of sodium without severe adrenal insufficiency symptoms before discharge from neonatal intensive care unit (NICU). The tandem mass spectrometry screening for congenital metabolic disease were normal including 17-OHP for congenital adrenal hyperplasia. The endocrinologic investigations including serum ACTH and cortisol were performed in both patients because of hyperpigmentation, mild electrolyte abnormalities although both patients did not show the severe adrenal insufficiency symptoms. Laboratory findings revealed normal 17-OHP, elevated ACTH (A, 4,379.2 pg/mL at 23 days of life; B, 11,616.1 pg/mL at 29 days of life), and high plasma renin activity (A, 49.02 ng/mL/hr; B, 52.7 ng/mL/hr). However, the level of plasma cortisol before treatment were normal (8.71 ┬Ąg/dL initially, 7.11 ┬Ąg/dL in 2nd sample) in patient A, but low (1.5 ┬Ąg/dL) in patient B. The results of endocrinologic investigation are shown in Table 1.
The enlargement adrenal glands in both patients were not remarkable on the abdominal ultrasound soon after birth. Moreover, hyperplasia of the adrenal glands was not observed on abdominal computed tomography (CT) performed at 11 months of age in both patients.
Patient A showed slightly decreased to normal sodium level (132 to 139 mmol/L) not to need sodium supplementation during hospitalization and she was discharged from NICU at 22 days of age. Discharge of patient B was delayed for correction of hyponatremia (129 to 133 mmo/L) and further evaluation for skin hyperpigmentation. She was treated with oral supplementation of sodium (0.8 to 0.9 mmol/kg) at 11 to 35 days of age, and she discharged from the hospital with normal sodium level after tapering off sodium supplementation at 37 days of age.
Patient A was admitted at 38 days of age with adrenal insufficiency symptoms of poor oral intake, vomiting, lethargy and dehydration. Clinical findings of patient A were suggestive of CLAH and prompted us to order a gene analysis in both twins. Chromosome analysis of both patients revealed normal 46, XX karyotype. The result of gene analysis of StAR revealed a heterozygous condition for c.544C>T (p.R182C) in exon 5 and c.722C>T (p.Q258X) in exon 7 (Fig. 1). Patient B started corticosteroid supplementation before manifestation of severe salt losing symptoms, except hyponatremia, but she was also diagnosed with CLAH with the same StAR gene mutation with patient A. The results of gene analysis of parents showed heterozygous for the mutations in the StAR gene; the heterozygous gene mutations; c.772C>T in the mother, and c.544C>T in the father, were documented.
They showed suboptimal levels of hormone despite of administration of 20 mg/m2/24-hr of hydrocortisone, dose of hydrocortisone was increased up to 30 mg/m2/24-hr. Both patients have been under steroid supplementation (fludrocortisones 0.2 mg daily in 2 divided doses, and hydrocortisone 12 mg daily in 3 divided doses) at 14 months of age. They showed normal development except retarded physical growth, and hormones and electrolyte levels on follow up are shown as Table 1. Body weight was 8.8 kg (3 to 10 percentile) in patient A, 9.4 kg (10 to 25 percentile) in patient B, body length was 73.7 cm (3 to 10 percentile) in patient A, 75.4 cm (10 to 25 percentile) in patient B at 14 months of age. They had been regularly followed.
Discussion
CLAH is an autosomal recessive disorder that is caused by a mutation in the StAR gene1,2) or mutations in the cholesterol side chain cleavage enzyme (CYP11A) in a rare case3). The defect in StAR dependent cholesterol transfer into mitochondria in the adrenal gland, mediated by the StAR protein resulted in accumulation of cholesterol esters in the adrenal gland and marked adrenal hyperplasia4). Patients with CLAH showed clinical history of adrenal insufficiency (salt loss, dehydration, vomiting, and lethargy) and electrolyte abnormalities (hyponatremia and hyperkalemia) within few months after birth and low level of adrenal hormones (cortisol, aldosterone, and dehydroepiandrosterone-sulfate), and testosterone and high ACTH level4). The patient who showed clinical findings of congenital adrenal hyperplasia but with normal range of 17-OHP should be considered the possibility of CLAH.
In this case, patient A showed normal cortisol level repeatedly before treatment, atypical case with detectable level of cortisol was reported previously5,6). Cortisol could be detected even in CLAH patient before enough accumulation of lipid to destroy the residual steroidogenesis of the adrenal gland in CLAH1). Although patient B was given the amount of oral sodium that is less than the daily requirement, in addition to steroid supplementation that was not required before discharge from the NICU, but patient A showed salt losing symptoms as early as one month of age despite normal cortisol level at initial. We assume that delayed manifestation of adrenal crisis in patient B despite of higher level of ACTH, and lower level of cortisol than patient A could result from sodium supplementation in NICU. Chen et al.7) also reported that different spectrum of clinical manifestations patients who belong to the same ethnic group with the same mutation.
Lipid droplet accumulation in the cytoplasm of adrenocortical cell2) works as the 2nd hit of the loss of steroidogenesis in the adrenal gland7) following mutation in the StAR gene as a 1st hit. Accumulated lipid work as toxic material in tissue and cause the further reduction in hormone synthesis independent of the StAR gene mutation1,2). Enlargement of the adrenal gland was also caused by accumulation of lipid droplet and it could be detected by radiologic images including ultrasound and CT in the early newborn stage8,9). In patients with CLAH, adrenal enlargement and increased echogenicity of adrenal gland could be seen on the abdominal ultrasound8,9). Ogata et al.8). reported the usefulness of CT for early diagnosis of CLAH, but no enlargement of the adrenal gland was observed in this case at 11 months of age as well as soon after birth. Although echogenicity of adrenal gland decreased during steroid replacement, the size of adrenal gland was independent with ACTH level and enlargement of gland persisted for 1year despite of steroid replacement in CLAH, it is differ from the response of congenital adrenal hyperplasia caused by 21-hydroxylase deficiency9). Some of cases with p.Q258X mutation also reported no adrenal gland enlargement despite of increased ACTH level (769 pg/mL)2), normal size of adrenal gland were observed in both patients from soon after birth in this study.
In patients with CLAH, the presentations with adrenal crisis symptoms were usually showed within the first 2 months of life, but late presentations were also reported as late as 10 to 14 months with the presence of residual StAR activity7). Bhangoo et al.2) reported a case with a homozygous missense mutation of c.544C>T (p.R182C) in exon 5 in Yemenian and genotypic male patients showed delayed presentation of adrenal crisis after 5 months of age. Therefore, the patient with homozygous for p.R182C mutation showed severe electrolyte imbalance (sodium, 114 mEq/L; potassium, 8 mEq/L), hypoglycemia (20 mg/dL), and neurologic abnormalities including frontal and temporal atrophy on brain magnetic resonance imaging, hypotonia and developmental delay2). Patient A was a heterozygous for p.R182C/p.Q258X mutation and adrenal crisis symptoms developed early of neonatal period around 1 month of age. In patients with transition arginine at codon 182 to other amino acid reported late onset of symptoms and mild symptoms1,2,7), but some of them showed symptoms early7). Mutation of p.Q258X resulted in premature stop codon and loss of StAR activity with formation of truncated protein10). Although there was no report functional study of p. R182C, failing formation of the cholesterol biding pocket was described in p.R182H2,7).
We report on the first case of the mutation of p.R182C in exon 5 of the StAR gene in Korea. To the best of our knowledge, mutation of Gln258* (p.Q258X) in exon 7 in the StAR gene was the most common mutation in Japanese and Korean patients1,7), the mutation of p.R182C in exon 5 in this twin is the first case in Korean patients.