Introduction
Somatic growth is dependent on an increase in both cell size and number. Human growth comes from a complex interplay of various factors including genetic backgrounds and environmental influences. However, complex mechanisms involved in the regulation of somatic growth remains to be completely understood particularly in the molecular level of genetic field. Overgrowth refers to a condition characterized by extreme physical size and stature including tall stature or generalized/localized overgrowth of tissues1). The condition originates during infancy, childhood or adolescence while epiphyseal growth plates remain open. Among various conditions showing overgrowth, genetic overgrowth syndrome refers to a nonhormonally mediated overgrowth condition which can accompany increased height and/or head circumference, various degrees of mental retardation, or physical dysmorphisms in children1). It is clearly distinguished from pituitary gigantism which is caused by excess of growth hormone. However, there are overlaps in clinical and molecular features between overgrowth syndromes, thus making a specific diagnosis is often difficult.
This paper reviews clinical characteristics and molecular basis of typical genetic overgrowth syndromes, focusing on Sotos syndrome (OMIM#117550) and Beckwith-Wiedemann syndrome (OMIM#130650).
Sotos syndrome
1. Clinical characteristics
Sotos syndrome (SS, OMIM#117550), also known as cerebral gigantism, is a prenatal and postnatal overgrowth syndrome characterized by excessive growth resulting in tall stature and macrocephaly, distinctive craniofacial features, and developmental delay. These three cardinal features are each present in over 90% of cases with SS2,3). Since the first description by Sotos et al.4) in 1964, hundreds of cases have been reported to date, and the estimated incidence is 1/15,000-1/20,0001).
The typical overgrowth pattern of SS starts prenatally, resulting in higher mean birth length and weight5). Pronounced postnatal growth is obvious in the first 6 years of life, consistently displaying height above the 97th percentile6). However, the final adult height is usually within the upper normal range due to accompanied bone age advancement6).
A characteristic facial appearance consists of a high and broad forehead, sparse fronto-temporal hair, malar flushing, down-slanted palpebral fissures and a pointed chin7). The head circumference is increased above the 97th percentile in most SS patients, and it is thought to be the most consistent indicator of SS at any age5).
The majority of SS patients have some degree of developmental delay/learning disability. Achievement of developmental milestones such as walking and speech is commonly delayed. However, most patients have mild to moderate intellectual impairment, and the severity is very broad, ranging from intelligence quotient below 30 to above 1002).
Besides, there are other commonly associated features including a history of neonatal jaundice and feeding difficulty, variable types of cardiac and renal anomalies, seizure, scoliosis, strabismus, attention deficit hyperactivity disorder, nonspecific abnormal brain image findings such as ventriculomegaly and corpus callosum hypoplasia. Patients with overgrowth syndromes including SS have higher risks for the development of neoplasias, particularly in their childhood. In SS patients, the frequency of tumor development has been reported to be 2-7%8,9), and the relative risk of certain malignancies including neural crest tumors, saccrococcygeal teratomas and some hematological malignancies is increased2). However, routine screening of tumor development is not a standardized recommendation.
2. Molecular and genetic basis
In 2002, the nuclear receptor set domain containing protein 1 gene, NSD1, on chromosome 5q35 was identified as a causing gene of SS10). SS is caused by haploinsufficiency of NSD1 in 60-90% of clinically diagnosed SS patients and can be transmitted in an autosomal dominant manner, although more than 95% of patients gain the disease from de novo mutation11).
The NSD1 gene consists of 23 exons and encodes multiple functional domains, including the SU(VAR)3-9, E(Z), tirthorax (SET), SET-associated domains, which mediate histone methyltransferase activity, five plant homeo-domains implicated in chromatin regulation, and two proline-tryptophan-tryptophan-proline domains that may mediate protein interactions3). NSD1 is expressed in several tissues including the brain, kidney, skeletal muscle, spleen, and thymus12). Although the exact role of the NSD1 protein has not been identified, the presence of two different ligand binding domains suggests that NSD1 enables the regulation of both negative and positive transcription13).
Several reports have demonstrated NSD1 abnormalities in patients with Sotos syndrome. NSD1 abnormalities include microdeletion of 5q35, encompassing the entire NSD1 deletions and mutations within the NSD1 gene (intragenic mutations). To date, over 300 different mutations associated with Sotos syndrome have been identified, including gross deletions, small indels, point mutations, and splice site mutations (Human Gene Mutation Database, http://www.hgmd.org/). Notably, 5q35 microdeletions are more frequently (approximately 50%) found in Japanese patients with Sotos syndrome10), whereas 5q35 microdeletions are uncommon (<15%) in patients in other areas of the world3)(Fig. 1). Recently, the first report of SS in Korea was published14), and this study documented that 53% of patients had a 5q35 microdeletion, a result that is very similar to that in Japan. Moreover, NSD1 abnormalities have been delineated in up to 90% of non-Japanese patients with Sotos syndrome, whereas approximately 30% of Japanese and Korean patients do not have NSD1 abnormalities14,15). 5q35 microdeletions can be detected by fluorescence in situ hybridization (FISH), multiplex ligation dependent probe amplification (MLPA), or array comparative genomic hybridization methods (array CGH), and NSD1 intragenic mutations can be identified by the direct sequencing method using the patient's genomic DNA.
However, the exact mechanism of such an interracial difference has not been clearly explained to date. Patients with a microdeletion tend to have certain congenital heart and/or urogenital anomalies, more severe mental retardation, and shorter stature than those with NSD1 intragenic mutations16).
Beckwith-Wiedemann syndrome
1. Clinical characteristics
Beckwith-Wiedemann syndrome (BWS, OMIM#130650) is the most common genetic overgrowth syndrome, with an estimated incidence of 1/13,70017). However, the actual incidence may be higher than the estimated incidence, considering the broad clinical spectrum of BWS18).
This syndrome is characterized by prenatal and postnatal overgrowth, polyhydramnios, abdominal wall defects including omphalocele, macroglossia, and visceromegaly of abdominal organs, hemihyperplasia, neonatal hyperinsulinemic hypoglycemia, ear creases and pits, cleft palate, and predisposition to embryonal tumors19). These clinical features are quite variable, and there are no absolute criteria for a clinical diagnosis to date. Although intelligence is usually normal, mild to moderate developmental delay can be found in patients with a history of hypoglycemia and airway problems19).
Because of the well documented association between BWS and increased risk of embryonal tumor development, early diagnosis of BWS is important in the clinical setting. Wilms tumor and hepatoblastoma are most commonly accompanied, and various malignancies including neuroblastoma, rhabdomyosarcoma, adrenocortical carcinoma, and benign tumors have als o been reported in BWS patients20,21). The published lifetime incidence of embryonal tumors in BWS patients varies between 4 and 21% (mean, 7.5%), and most of the tumors are present in the first 8 years of life19). Therefore, tumor surveillance is particularly targeted at detecting Wilms tumor and hepatoblastoma, and abdominal ultrasonography during infancy and early childhood is highly recommended to all BWS patients22). Tumor surveillance may reduce treatment-related morbidities if tumors can be detected at early stages.
Various protocols for tumor surveillance have been proposed20,23,24). Although there are no established consensus guidelines for tumor surveillance in BWS, most previous studies suggest abdominal ultrasonography every 3-6 months to the age of 8 years and measuring serum alpha fetoprotein every 2-3 months until 4 years20,23,24).
2. Molecular and genetic basis
The heterogeneous molecular basis for BWS has been studied since the mapping of chromosome 11p15 as the causative locus for BWS by Waziri et al.25) in 1983. However, due to the genetic complexity in BWS, the molecular diagnosis of BWS is still difficult, and the diagnosis of BWS mainly relies on clinical features.
BWS is caused by dysregulation of imprinted growth regulatory genes on chromosome 11p1526). In contrast to most autosomal genes, only one allele of either the paternal or maternal allele in each imprinted gene is expressed normally. It is a parental origin-specific manner. Abnormalities in the imprinting process of a number of growth regulatory genes within the two domains on 11p15 lead to dysregulation of prenatal and postnatal growth and cause BWS19).
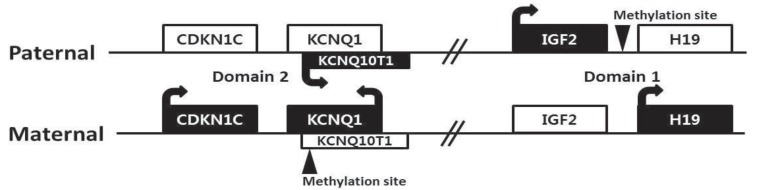
There are four major genes (IGF2, H19, CDKN1C, and KCNQ10T1) associated with genomic imprinting in two imprinting domains (imprinting domain 1 and 2) on 11p15, and expression of these genes in each domain is cis-regulated by imprinting centers 1 and 2, respectively (Fig. 2). IGF2 and H19 are located in domain 1, and CDKN1C and KCNQ10T1 are located in domain 2. Normally, IGF2 is a paternally expressed gene and encodes insulin-like growth factor 2, which is the major growth factor in utero that promotes prenatal growth. H19 is a maternally expressed gene and encodes an untranslated mRNA, which may function as a tumor suppressor and a growth restrictor27,28). CDKN1C is a maternally expressed gene and encodes a cyclin-dependent kinase inhibitor that negatively regulates cell proliferation29). KCNQ10T1 is a paternally expressed gene and encodes untranslated mRNA, which may act as a negative regulator for CDKN1C expression30). Therefore, paternally expressed genes (IGF2 and KCNQ10T1) promote growth particularly in utero, and maternally expressed genes (H19 and CDKN1C) restrict growth, opposing the effect of paternally expressed genes.
In BWS, alterations of these imprinting processes in the genes of domain 1 and 2 can be molecularly identified in approximately 70% of patients by various kinds of methylation studies19). Approximately 50% of patients show hypomethylation of domain 2 in the maternal allele, causing expression of maternal KCNQ10T1. Increased expression of IGF2 occurs in approximately 25% of patients via paternal uniparental disomy (20%) or hypermethylation of domain 1 in the maternal allele (5%). The broad clinical spectrum of BWS is likely associated with the heterogeneity of these underlying genetic mechanisms. Furthermore, approximately 5% of patients have intragenic mutations in CDKN1C of maternal allele. However, more than 20% of BWS patients still remain with undetermined molecular causes19). Methylation-specific MLPA is currently the most robust method to confirm BWS, and it can detect methylation defects of domain 1 or 2 (55%), paternal uniparental disomy (20%), and microdeletion/microduplication involving domain 1 and/or 2 (rare). Cytogenetically detectable abnormalities using conventional karyotyping and FISH analysis are found only less than 1% in BWS patients. Sequence analysis of the CDKN1C gene can identify intragenic mutations (5%)31).
Interestingly, the 11p15 imprinting region is also associated with Russell-Silver syndrome (RSS, OMIM#180860), which is a typical growth retardation syndrome32,33). In contrast to BWS, silencing paternally expressed genes (IGF2 and KCNQ10T1) or enhancing maternally expressed genes (H19 and CDKN1C) leads to RSS34). Opposite epigenetic alterations in 11p15 result in opposite clinical features shown in BWS and RSS. Alterations in the imprinting process on 11p15 can be identified in approximately 50% of RSS patients32,33).
Conclusions
Human growth ensues from a complex interplay of genetic traits and environmental influences. In particular, basic studies have continued to clarify the molecular basis of growth disorders in the genetic field. Although the exact functions of the causing genes have not yet been completely understood, genetic overgrowth syndromes can be good models to clarify the complex basis of human growth and assist with the development of better-directed therapies in the future.