Highlights
· Congenital hypogonadotropic hypogonadism is a rare and treatable form of male infertility.
· The treatment strategies throughout life are very important and proper approaches can significantly improve the long-term quality of life.
Introduction
Puberty is one of the most significant changes in human development and is accompanied by the expression of secondary sexual characteristics, such as breast engorgement and testicular enlargement, achievement of adult height, and completion of fertilization ability [1]. These pubertal changes can be attributed to activation of the hypothalamic-pituitary-gonadal (HPG) axis. In the hypothalamus, gonadotropin-releasing hormone (GnRH) stimulates the pituitary gland to secrete luteinizing hormone (LH) and follicle stimulating hormone (FSH), eventually triggering testicular development in males as well as testosterone production and reproductive functions such as spermatogenesis [2].
Congenital hypogonadotropic hypogonadism (CHH) is characterized by complete or partial failure of pubertal development due to inadequate secretion of LH and FSH. Hypogonadotropic hypogonadism includes (1) hypogonadal hypogonadism associated with abnormal olfactory neuronal migration (anosmia/hyposmia), called Kallmann syndrome (KS); (2) hypogonadotropic hypogonadism with normal olfactory function (normosmic isolated hypogonadotropic hypogonadism, nIHH); and (3) acquired hypogonadism [3].
CHH is one of the few treatable diseases of male infertility, although men with primary testicular dysfunction show irreversibly diminished spermatogenic capacity [4-6]. The approach to CHH treatment is determined by goals such as developing only virilization or inducing fertility. In this review, we describe therapeutic options from different perspectives. To this end, we also reviewed the normal physiology of the reproductive axis.
Normal physiology of the reproductive axis
GnRH is a peptide consisting of 10 amino acids and is encoded by a gene located on chromosome 8 [7]. In the fetus, at 14 weeks of gestation, neurons containing GnRH exist in the hypothalamus, As the hypothalamus-pituitary portal system develops, GnRH in the hypothalamus reaches the gonadotroph in the pituitary gland. At 20 weeks of pregnancy, LH and FSH are secreted by the pituitary gland. Thereafter, secretion of gonadotropin increases by GnRH stimulation until the middle of pregnancy and then decreases until birth when LH and FSH are inhibited in the upper central nervous system. During labor, the concentration of gonadotropins is lower than that in the second trimester but is still relatively high [8]. Even after birth, gonadotropins are secreted intermittently in large amounts, and the HPG axis remains active during the first 6 months of life, with increased secretion of gonadotropin approaching pubertal level. This so-called "mini-puberty" is a significant clue to diagnosis of CHH among males exhibiting cryptorchidism with or without micropenis, as low serum testosterone and LH levels can be used to identify congenital GnRH deficiency. [9].
The HPG axis is reactivated at pubertal onset, with reappearance of pulsatile GnRH release. In men, just before entering puberty, intermittent LH secretion occurs mainly during sleep; as puberty progresses, the frequency and amplitude of GnRH secretion increase, as does daytime secretion. This is due to activation of kisspeptin signals, which direct GnRH neurons to control pulsatile GnRH release. This occurs via elevation of LH and FSH levels through the pituitary, with downstream activation of sex steroids, including estrogen and testosterone [10]. LH stimulates maturation of interstitial Leydig cells, which secrete testosterone and insulin-like factor 3. When LH binds to the cell membrane receptor of Leydig cells, the level of cAMP increases through adenylate cyclase, which stimulates protein kinases to convert cholesterol to pregnenolone by P450scc, the first step in testosterone production [11]. In addition to direct secretion from the testes, testosterone is produced in small amounts by conversion of androstenedione secreted by the testes and adrenal glands. Secretion of testosterone induces and maintains spermatogenesis in Sertoli cells. In addition, FSH stimulates proliferation of immature Sertoli cells, which are indispensable for development of the seminiferous tubule, where spermatogenesis occurs. Seminiferous tubules account for approximately 90% of the testicular volume [12]. Testicular volume is an important predictor of fertility in males with CHH [13]. FSH-induced proliferation of immature Sertoli cells determines the final seminiferous tubule length. Therefore, both FSH and testosterone levels are essential for normal spermatogenesis [14].
Interestingly, Sertoli cells do not express androgen receptors during mini-puberty [9]. Thus, despite the LH-induced high intratesticular testosterone level, Sertoli cells remain immature. As puberty progresses, testosterone production by Leydig cells increases and leads to maturation of Sertoli cells and onset of spermatogenesis [15]. In particular, the level of testosterone secreted by Sertoli cells is much higher than that in the peripheral circulation, which is required for spermatogenesis. This has been demonstrated in studies of men with CHH, where FSH was shown to bind to LH-induced testosterone and stimulate spermatogenesis, but not FSH or exogenous testosterone [16].
Congenital hypogonadotropic hypogonadism
CHH is characterized by absent or incomplete sexual development and/or infertility due to GnRH deficiency. Unlike acquired diseases caused by damage to the pituitary gland and hypothalamus, which are characterized by deficiencies in multiple pituitary/hypothalamic hormones, CHH is characterized by isolated GnRH deficiency. GnRH deficiency is more common in boys than in girls and occurs in 1 in 4,000– 10,000 men [17].
Patients with CHH typically present with absent puberty in adolescence or early adulthood. Adolescent boys tend to have prepubertal testicular volumes less than 4 mL with undervirilization. The biochemical profile of CHH consists of low testosterone level and inappropriately low or normal serum gonadotropin level. In addition, CHH shows a spectrum of severities along with a number of associated phenotypes that appear at variable rates, including cleft palate, labia majora, renal malformation, nystagmus, and bimanual synkinesia (mirror movements) [18].
Causes of congenital hypogonadotropic hypogonadism
Hypogonadotropic hypogonadism is often caused by genetic mutations. Technological advancements in genetics have had a profound impact on the research and diagnosis of noncommunicable diseases [19]. Over the last 30 years, several research groups worldwide have used a combination of clinical investigational strategies and genetic approaches to elucidate the genetic complexity underlying CHH and link its phenotypic variations to specific genetic and biological causes [20]. The ability of next-generation sequencing to query the entire genome with increasing speed and accuracy is identifying a surprising number of rare sequence variants of unknown significance in both known and novel CHH genes. Moreover, these technical advancements in genetic testing have been accompanied by new challenges in sequence interpretation [21]. To date, mutations in more than 35 genes have been shown to be associated with CHH. However, mutations in many different genes can only explain approximately 40% of the causes of CHH, with the majority of CHH patients being genetically uncharacterized [22-24]. The mode of inheritance is X-linked, autosomal dominant, and autosomal recessive; however, sporadic occurrences are more common. Moreover, the genetics of CHH are complex; even if hypogonadal hypogonadism is caused by one single mutation, the phenotype may vary [25].
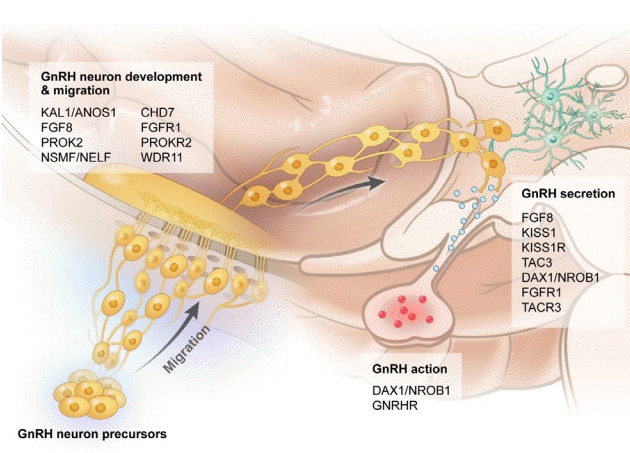
The first gene associated with KS is KAL1 in Xp22.3 [26]. In a normal fetus, neurons that generate GnRH originate in the medial epithelial cells of the olfactory placode during the early stages of development and move to the hypothalamus. This shift is regulated by the KAL1 gene [25]. Anosmin 1 produced in the KAL1 gene is an extracellular matrix glycoprotein involved in the growth and migration of GnRH neurons and olfactory nerves during early embryogenesis [27]. In typical KS, one or both olfactory bulbs are invisible. An magnetic resonance imaging scan can confirm size reduction or aplasia of the olfactory bulb. The degrees of olfactory and gonadal dysfunction are diverse, and even within a family, the degree of abnormality may vary. Other genes such as NSMF, FGFR1, FGF8, FGF17, IL17RD, PROK2, PROKR2, HS6ST1, CHD7, WDR11, SEMA3A, TUBB3, and SOX10 are associated with KS-encoded proteins that act cooperatively with anosmin 1, which is related to migration of GnRH neurons [22,26,28].
In addition, mutations in genes related to GnRH secretion, such as GnRH1, KISS1, KISS1R, TAC3, TACR3, LEP, and LEPR, cause nIHH with normal olfactory function [26,29]. However, mutations in genes such as NSMF, FGFR1, FGF8, FGF17, IL17RD, PROK2, PROKR2, HS6ST1, CHD7, WDR11, and SEMA3A can cause both KS and nIHH but retain normal olfactory function (Fig. 1) [30].
Treatment of CHH
1. Infants
The focus of most treatments in affected boys with micropenis or cryptorchidism is appropriate testicular descent and penile growth. Cryptorchidism has a negative effect on future fertility potential. Therefore, early surgical correction is important, and current treatment guidelines recommend surgical intervention within 6 months to 1 year of age. The micropenis should be treated early using short-term, low-dose testosterone to induce penile growth. Testosterone therapy is usually initiated within the first 3 years of life. A common treatment regimen is intramuscular administration of 25 mg of testosterone esters (testosterone enanthate or cypionate) every 3 weeks for 3 months. Adverse reactions, including virilization or secondary sexual development, were minimal due to the short duration of treatment.
Gonadotropins such as LH and FSH can be used to increase penile length in infant boys with micropenis. Main et al. [31] first reported the effects of short-course recombinant LH and FSH treatment in a boy with CHH. The patient was treated with a subcutaneous injection of recombinant human LH and FSH twice a week from 7.9 to 13.7 months of age. With this treatment, penile length increased from 1.6 cm to 2.4 cm and testicular volume increased by 170%. In addition, the levels of LH, FSH, and inhibin B increased. The treatment was well-tolerated, even though certain adverse reactions, such as increased body hair, increased pigmentation, and intermittent vomiting, were noted. In another study by Bougnères et al. [32], 2 neonates with CHH and one with congenital hypopituitarism were treated with recombinant LH and FSH via a pump for 6 months starting from the newborn period. This treatment led to increase penile length from 8 to 30 mm and 12 to 48 mm, respectively, as well as an increasement of testicular volume (0.57 to 2.1 mL and 0.45 to 2.1 mL, respectively). A recent study reported the effects of recombinant gonadotropin infusion in 4 males with CHH [33]. Serum testosterone level, inhibin B, and penile length were significantly increased for a mean duration of 4.2±1.2 months. This treatment was well-tolerated and resulted in no adverse events. These studies suggest that gonadotropin treatment may be beneficial for testicular development and future reproductive functions. FSH treatment stimulates the proliferation of immature Sertoli cells and growth of seminiferous tubules in males with CHH before pubertal induction. However, these treatments were limited by the small sample sizes. Further investigations with a larger number of patients and randomized controlled trials are needed to examine the effectiveness of gonadotropin treatment in male patients with CHH.
2. Adolescence and adulthood
Delayed puberty is associated with significant psychosocial impairments, including low self-esteem, social withdrawal, low academic achievement, and higher rates of substance abuse disorders. Therefore, the treatment goals are to achieve virilization, normal sexual function, fertility, optimal adult height, and normal psychosocial development.
1) Induction of male secondary sexual characteristics
Testosterone injection is the mainstay treatment for induction of secondary sexual development. In this treatment approach, testosterone esters, such as testosterone enanthate or cypionate, are administered through intramuscular injections every 4 weeks. A low dose of testosterone (50 mg) is usually started at a bone age of at least 12 years and gradually is increased to the full adult dose (250 mg) over 24–36 months [34]. This pubertal induction regimen seeks to replicate the typical tempo of puberty and maximize adult height. Testicular volume and serum LH, FSH, and testosterone evels are monitored every 6 months, and the dose of testosterone can be gradually increased to the full adult dose depending on the monitoring results. The adverse reactions caused by testosterone treatment include erythrocytosis, premature closure of the epiphysis, and occasional pain or erythema at the injection site [35].
2) Induction of male fertility
CHH is a rare and treatable disease that is associated with infertility. However, exogenous testosterone treatment does not induce testicular growth or spermatogenesis, since exogenous testosterone suppresses the production of endogenous testosterone required for spermatogenesis. Therefore, pulsatile GnRH treatment, hCG monotherapy, or FSH and hCG combination therapy can be considered to induce fertility in men with CHH (Table 1). Several predictors of fertility outcomes have been reported in CHH, regardless of the treatment regimen [36-40]. Testicular volume is a key predictor of fertility. Male CHH patients with prepubertal testes consistently have poorer fertility outcomes. In addition, a history of cryptorchidism is a poor prognostic factor for fertility. Warne et al. [36] reported that prior testosterone treatment was a negative prognostic factor for fertility outcomes. However, this effect was not observed in a subsequent large meta-analysis [13]. Thus, the effect of prior testosterone treatment on fertility remains questionable.
3) Pulsatile GnRH treatment
Since patients with CHH are deficient in GnRH, administration of GnRH may be the most ideal treatment method. However, because secretion of physiological GnRH is pulsatile, continuous GnRH injection leads to desensitization of the GnRH receptor, which inhibits gonadotropin secretion. Therefore, GnRH (25 ng/kg every 2 hours, titrated for serum testosterone level) should be administered in a pulsatile fashion through an infusion pump [39]. Several studies have reported that pulsatile GnRH infusion successfully induces puberty and fertility, with approximately 80% of men being able to achieve spermatogenesis during long-term treatment [41-43]. However, this regimen may be limited because of the inherent difficulty in pulsatile GnRH administration, and it does not show a significant difference in fertility outcome in comparison with gonadotropin treatment [44].
4) hCG monotherapy
hCG monotherapy has been used as a treatment option to induce fertility, but this therapy is only suitable for men with the mildest form of the phenotypic spectrum, such as adultonset hypogonadism, larger testicular size, or no history of cryptorchidism [45]. Fertility is usually achieved after 3–9 months [46].
In clinical practice, hCG is usually started at 3,000–5,000 IU per week divided into 2–3 self-administered subcutaneous injections. In patients with testicular volume less than 4 mL or a history of cryptorchidism, the rate of treatment failure is high, even with a longer treatment duration [37]. If the sperm count is sufficient after 3–6 months of treatment, FSH can be added to achieve better outcomes. Common side effects include erythrocytosis and gynecomastia. Serum testosterone, hematocrit, and estradiol levels were monitored every 4–6 weeks to titrate the dose of hCG.
5) Combined gonadotrophin treatment
Depending on the severity of the disease, including testicular volume or history of cryptorchidism, a combination of FSH and hCG may be required to induce fertility in men with hypogonadism. Commonly available gonadotropin preparations are highly purified urinary gonadotropins and recombinant FSH [47]. In addition to hCG treatment, FSH is practically initiated at a dose of 75 IU every other day by subcutaneously self-administered injection and increased up to 150 IU 3 times per week to achieve a serum FSH concentration in the physiological range of 4–8 IU/L [48]. A recent meta-analysis reported the effects of hCG monotherapy or in combination with FSH [13]. Gonadotropin stimulation was beneficial in 75% of the cases, which resulted in a final mean sperm count of 5.92×106/mL, which is sufficient to achieve fertility despite being at a subphysiological level. These data indicate the beneficial synergistic effect of combined FSH and hCG treatment.
6) Sequential gonadotrophin treatment: FSH pretreatment followed by combined FSH and hCG therapy
Men with CHH have significantly reduced spermatogenic capacity because of the absence of FSH-stimulated Sertoli and germ cell proliferation. Therefore, pretreatment with FSH, followed by combined hCG and FSH therapy, may be an optimal therapeutic strategy for inducing fertility in patients with CHH before exposure to hCG or GnRH-induced endogenous LH [49]. Dwyer et al. [50] reported the results of 24 months of GnRH therapy after 4 months of FSH pretreatment (n=7) in comparison with pulsatile GnRH therapy alone (n=6) in CHH patients with immature testes (testicular volume < 4 mL). The FSH treatment group showed significantly increased inhibin B level to the normal range and an approximately 2-fold increase in testicular volume. All 7 patients who received FSH pretreatment developed sperm in ejaculation, in comparison with 4 of the 6 patients in the GnRH monotherapy group, and showed an increasing tendency in maximum sperm count.
Conclusion
CHH is a rare and treatable form of male infertility. Men with CHH usually have a poor prognosis because of severely early impaired fetal testicular development. Thus, treatment strategies throughout life are very important, and proper approaches can significantly improve the long-term quality of life. To date, no clear protocol has been established for treatment of men with CHH. Further studies, including randomized controlled trials with large patients, are needed to establish treatment guidelines for males with CHH.