Highlights
Myhre syndrome is a rare autosomal-dominant disorder characterized by short stature, intellectual disability, skeletal anomalies, restricted joint mobility, distinctive facial dysmorphism, and deafness. We report the first Korean case of genetically-confirmed Myhre syndrome in Korea.
Introduction
Myhre syndrome (MS; OMIM 139210) is a rare autosomal-dominant disorder characterized by short stature, intellectual disability, skeletal anomalies, restricted joint mobility, distinctive facial dysmorphism, and deafness [1]. The skeletal anomalies include thickened calvarium, brachydactyly with cone-shaped epiphyses, shortened tubular bones, hypoplastic iliac wings, and large pedicles of the vertebrae [2]. Facial dysmorphism consists of maxillary hypoplasia, prognathism, short palpebral fissure, short philtrum, and small mouth. Additional multisystem involvement is reported, including in the cardiovascular (congenital heart defects, pericardial effusion, constrictive pericarditis, restrictive cardiomyopathy, and hypertension), respiratory (choanal stenosis, laryngotracheal stenosis, and restrictive pulmonary disorder), and gastrointestinal (pyloric stenosis and duodenal stricture) systems [3]. Patients with MS can develop various long-term and life-threatening complications [4]. Recently, SMAD4 was identified as the major gene responsible for MS. Most patients have missense mutations at a single codon site (Ile500) of the SMAD4 gene [1]. To date, there have been 79 reported cases worldwide [5]. In this study, we report the first Korean case of MS.
Case report
The patient was a 13-year-old female who was born to a 37-year-old female and a 38-year-old male. She was delivered vaginally at 41 weeks of gestation and weighed 2.6 kg (-1.6 standard deviation score [SDS]). She had a healthy brother, and there was no family history of short stature, mental retardation, or genetic disorders. Her father's height was 178 cm (1.2 SDS), and her mother's height was 161 cm (0.5 SDS). At the age of 5 months, she was found to have a patent ductus arteriosus (PDA) and received PDA ligation.
Her early developmental milestones were below normal limits. While attending preschool, she was found to have learning disabilities, delayed motor development, and impaired social interactions. She was nonverbal in preschool and diagnosed with selective mutism when she was 5 years old. She was referred to our neuropsychiatric department when she was 6 years old. A psychological test estimated her intelligence quotient and mental age to be 64 and 5.5 years, respectively. Her height had always remained below the third percentile.
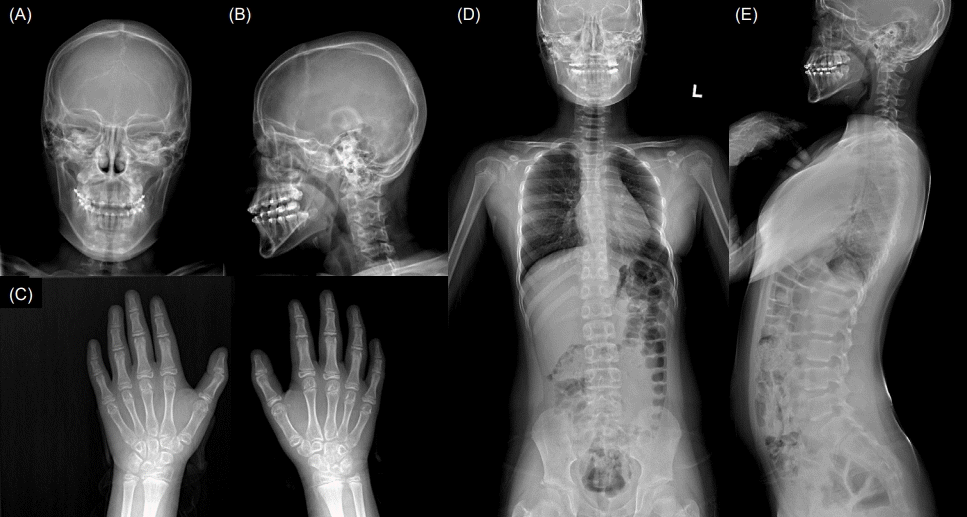
At 8 years old, she was referred to the pediatric endocrinology clinic due to her short stature. On physical examination, her height was 115.5 cm (-2.1 SDS), and her weight was 26.0 kg (0 SDS). She had a short neck, prognathism, blepharophimosis, clinodactyly, and a shield chest (Fig. 1).
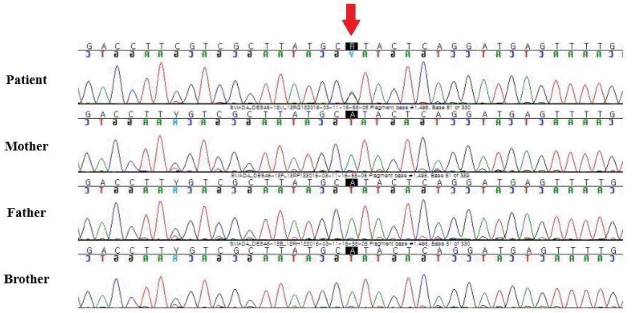
Endocrinological tests showed normal insulin-like growth factor-1 level and thyroid function. The results of chromosome analysis were normal (46,XX). No pathologic findings were identified in mutation analysis of the PTPN11 gene for Noonan syndrome. The results of brain magnetic resonance imaging were normal. Skull radiographs showed a thickened calvarium and prognathism (Fig. 2A, B), a hand radiograph showed clinodactyly and bone age appropriate for chronological age (Fig. 2C), and a whole-spine radiograph showed large vertebral pedicles and mild scoliosis (Fig. 2D, E). Clinical exome sequencing was performed and revealed a c.1498A>G (p.Ile500Val) variant in the SMAD4 gene, which was confirmed by Sanger sequencing. Further, familial genotyping (farther, mother, and younger brother) was performed by Sanger sequencing. According to the genetic test results (Fig. 3), the detected variant was confirmed as a de novo variant. Moreover, it is an additional evidence of pathogenicity according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines (PM6: De novo [assumed] in a patient with the disease and no family history) [6].
Ophthalmologic examination, audiometric assessment, and laryngoscopy were performed to identify complications such as hypermetropia, farsightedness, astigmatism, strabismus, congenital cataract, hearing loss, and laryngotracheal stenosis. No complications were found.
When the patient was 8 years 11 months, her physical examination revealed a height of 117.0 cm (-2.0 SDS) and a weight of 27.1 kg (-0.4 SDS; body mass index=19.8 kg/m2, 1.0 SDS). Her breast development was Tanner stage II. A luteinizing hormone-releasing stimulation test showed a peak level of 5.8 IU/L. Her bone age was 10.5 years. These findings indicated central precocious puberty.
After receiving the diagnosis of central precocious puberty, she started gonadotropin-releasing hormone agonist (GnRHa). She simultaneously started recombinant human growth hormone (rhGH) treatment because she was born small for gestational age, and no catch-up growth had occurred. GnRHa treatment was discontinued after about 3 years, and rhGH treatment was discontinued at the age of 14 years due to her bone age of 15 years. After 4-year GnRHa and 5-year rhGH treatments, her final height improved from -2.0 SDS at 8 years 11 months (117.0 cm) to -1.6 SDS at 14 years (150.6 cm).
Discussion
MS is a rare developmental disorder affecting various systems over time [7]. Early diagnosis of MS is difficult because some of its features become apparent at school age [8]. Multifocal investigation is required to monitor potentially life-threatening complications.
MS shares some of its features with other syndromes. The differential diagnosis includes disorders like acromicric dysplasia, geleophysic dysplasia, and Weill-Marchesani syndrome. Acromicric dysplasia overlaps with MS in displaying short stature, thickened skin, and joint stiffness, but MS is distinguished by facial features, intrauterine growth restriction, mental retardation, and severity of muscular and cutaneous involvement, as well as more frequent cardiac anomalies and hearing loss [9]. Geleophysic dysplasia is a progressive disorder characterized by short stature, short hands and feet, and distinctive facial features and is associated with organomegaly, which is rare in MS [3]. Weill-Marchesani syndrome is characterized by short stature, brachydactyly, and joint stiffness, but it also features typical ocular disorders including microspherophakia, myopia, ectopia lentis, and glaucoma [10]. While SMAD4 mutations have been identified in MS, FBN1 mutations have been identified in acromicric dysplasia, ADAMTSL2, and FBN1 mutations in geleophysic dysplasia, and ADAMTS10, LTPBP2, and FBN1 mutations in Weill-Marchesani syndrome.
The patient in the present case exhibited typical features of MS, including short stature, intellectual disability, and facial dysmorphism. Identification of a SMAD4 gene mutation led to the diagnosis.
MS was first described in 1981, as a rare condition defined by a combination of the following symptoms: short stature, limited joint mobility, facial and skeletal dysmorphism, muscular hypertrophy, thick skin, variable intellectual performance, and ophthalmological and cardiovascular complications [4,11]. De novo heterozygous missense mutations in the SMAD4 gene have been identified as an underlying mechanism of the syndrome via disruption of the transforming growth factor-beta/bone morphogenetic protein (TGF-β/BMP) signaling cascade involved in embryonic development of connective tissue and the cardiovascular and central nervous systems [4,11,12]. SMADA4 is a central signaling component of the SMAD pathway that induces signals from TGF-β, a key mediator of fibrotic disease [13]. TGF-β ligands are drivers of extracellular matrix deposition, creating a concentrated pool of pro-fibrotic factors at the site of injury [14].
Typical morphological features of MS (including narrow palpebral fissures, prognathism, hypoplasia of the midface, and clinodactyly) were observed during physical examination of our patient. Radiological signs related to the syndrome were found (i.e., thick skull bones and mild scoliosis).
Although none of these were present in our patient, other symptoms commonly found in MS include ophthalmological and auditory impairments, such as cataracts, disordered retina, pseudo-papilledema, and refractory abnormalities such as hypermetropia and astigmatism, together with variably expressed hearing loss [11,15]. The majority of MS patients has delayed psychomotor development, autistic features, and variably expressed intellectual disability, all of which were present in our patient [9,16]. Missense mutations of the SMAD4 gene were among the de novo mutations in 200 genes important in neurodevelopmental disorders [17]. However, the impact of the detailed pathomechanism of mutant SMAD4 protein and TGF-β/BMP pathways on central neurological conditions is not known.
Over 70% of patients with MS have cardiovascular abnormalities, namely PDA, aortic or mitral valve stenosis, hypoplasia and/or stenosis of the abdominal aorta, pulmonary artery stenosis, pulmonary hypertension, and pericardial effusion [11,15]. Our patient had PDA and underwent PDA ligation.
Respiratory difficulties, including laryngo-tracheomalacia, bronchiolitis obliterans with organizing pneumonia, stenoses, and obstruction of both the upper and lower respiratory tracts such as that observed with hypertrophic adenoids, are common findings in MS and can lead to chronic progressive pulmonary inflammation [18]. Extrinsic interventions, such as intubation, often lead to exacerbation of existing stenoses due to defective tissue healing [19]. None of these respiratory difficulties were identified in our patient.
Sexual development problems are reported frequently in MS patients, including abnormal onset of puberty, premature menarche, and secondary amenorrhea in females and cryptorchidism and hypospadias in males [5]. While sexual development problems have been reported in many patients, little is known about the mechanism of SMAD4 variant disruption of the hypothalamic-pituitary-gonadal axis in MS cases [5]. Our patient was diagnosed with central precocious puberty at 8 years 11 months. The combined treatment of GnRHa and rhGH for children with central precocious puberty is recommended for patients with growth retardation and precocious puberty. Our patient received the combined treatment and showed improved final height.
MS is a rare genetic disorder that is caused by a SMAD4 mutation. The diagnosis should be considered when patients present with characteristic facial dysmorphism, joint contractures, brachydactyly, and short stature. Regular surveillance and anticipatory monitoring are necessary in MS because of associated life-threatening cardiovascular and respiratory manifestations [20]. Moreover, patients with MS can have a markedly abnormal fibroproliferative response to surgical intervention. Therefore, surgical intervention should be approached with extreme caution and with as little invasion as possible owing to the tendency for fibrosis development, which can cause significant morbidity and mortality.
In conclusion, we described the first Korean patient with MS, and the diagnosis was confirmed by the identification of a SMAD4 gene mutation.