Highlights
· Gamma-aminobutyric acid (GABA) may delay type 1 diabetes mellitus onset by promoting β-cell proliferation and modulating immune responses.
· GABA therapy can elevate insulin levels, inhibit β-cell apoptosis, and suppress T helper 1 cell activity against islet antigens.
· Clinical studies have shown no serious adverse effects of oral GABA, making it a promising preventative measure for high-risk pediatric populations.
Introduction
Type 1 diabetes mellitus (T1DM) is a severe autoimmune disorder resulting from autoantigen-specific T cells, leading to the elimination of pancreatic β cells that causes hyperglycemia due to insulin deficiency in the blood [1]. Predictions indicate a 60% to 107% increase in global T1DM cases by 2040 [2]. The highest prevalence of T1DM is found in children under 14 years of age, particularly those aged 4–6 and 10–14 years [3]. Managing T1DM complications and optimizing glucose levels are crucial. The current therapy involves lifetime insulin administration and lifestyle changes but has not entirely prevented complications, especially in children. A multicenter study with 10,000 participants reported only 13.1% achieved target glycated hemoglobin A1c (HbA1c) levels [4].
Gamma-aminobutyric acid (GABA) has immunomodulatory effects and induces β-cell regeneration,including the modulation of insulin secretion through GABA subtype B (GABAB) receptor stimulation [5,6]. GABA is orally available and safe for the central nervous system (CNS). GABA holds great promise in delaying T1DM onset in children, considering its minimal side effects and lack of serious adverse events during treatment [7,8]. With its beneficial immunomodulatory and β-cell regeneration effects, GABA represents a promising treatment option.
Type 1 diabetes pathophysiology
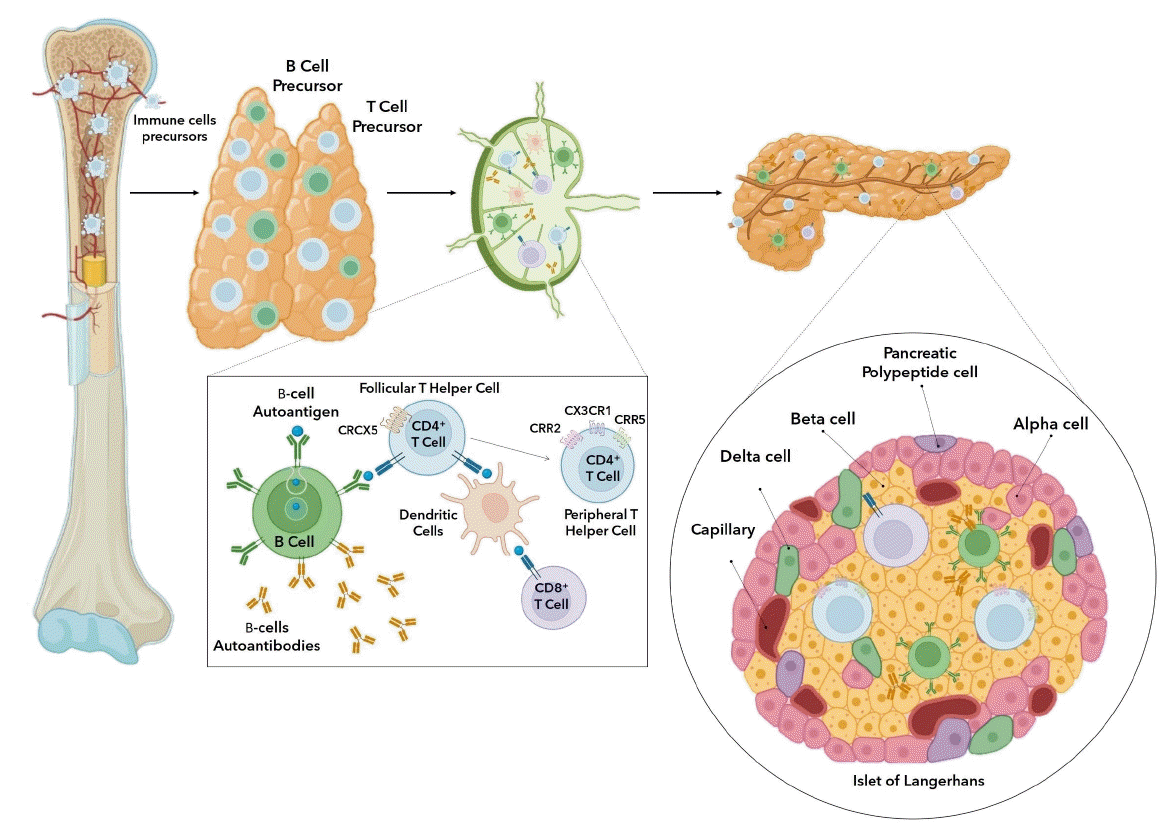
The pathogenesis of T1DM can be separated into a series of phases that starts with the identification of autoantibodies proceeding to cell death, dysglycemia, and hyperglycemiarelated symptoms [9]. The cause of cell-targeted autoimmunity involves a confluence of genetic and environmental variables that either initiate or facilitate the immune reaction against the cells [10]. The definitive cause of T1DM has not been determined; the variability of the condition is a challenging element when identifying disease-inducing variables. The numerous particular autoantibodies leading to autoimmunity are linked with diverse genetic susceptibility factors that reflect this variation [11,12]. T1DM is characterized by selective involvement of βcells without obvious pathogenic abnormalities of other Langerhans cells, such as α-cells, δ-cells, and pancreatic polypeptide cells [13]. High insulin autoantibody levels indicate early diabetes development in animals and young patients [14]. Therefore, mutations in the insulin gene promoter probably lead to a thymus with impaired central tolerance and the release of insulin-reactive T cells that direct the growth of autoreactive B cells [15]. Self-antigens secreted by damaged β cells are acquired by antigen-presenting cells (APCs), which then transport these materials to pancreatic lymphatic system and are passed on to autoreactive T lymphocytes. These T cells are uncontrolled components that are activated by genetically determined thymic deletion failures in combination with inadequacies in processes intended to potentiate peripheral immunological tolerance [16]. Following the inadequacies in central and peripheral tolerance, follicular T helper cells specific for islet antigens are generated in the pancreatic lymphatic system and assist autoreactive B cells in producing islet autoantibodies. Peripheral T helper cells generate CXCL13, which draws B cells towards inflamed islets, and interleukin (IL)-21, which enhances B-cell differentiation and leads to local autoantibody synthesis. Activated B cells generate cytokines and can operate as APCs to encourage increased immunological activation [17]. As a result of thymic plasma cell autoantibodies targeting certain medullary thymic epithelial cells for antibody-mediated apoptosis, the production of T cells is increased, bypassing negative selection and accelerating the course of T1DM [18].
B- and T-cell precursors are further developed in the thymus after formation in the bone marrow (Fig. 1) [19]. Beta cells and other islet cells produce type I interferon, such as interferon, which prompts the recruitment of immune cells. One of the initial cells to react are macrophages, which are also the major cell type that produces tumor necrosis factors (TNFs) [20]. Macrophages, acting as APCs, present autoantigens to the lymphatic system. Naive autoantigen-specific T lymphocytes in pancreatic lymph nodes identify islet molecular components from injured cells presented by APCs from pancreatic cells. CD4+, CD8+, and dendritic cells engage with activated B cells. T lymphocytes recognize autoantigens and eventually evade to the periphery [21,22]. Cell-specific T lymphocytes are activated by antigen presentation by dendritic cells and B cells. Autoreactive CD8+ and CD4+ T cells play significant roles in β-cell destruction by recognizing biomolecules of β-cell antigens such as precursor preproinsulin, tyrosine phosphatase-like insulinoma antigen, islet-specific glucose-6-phosphate catalytic subunit-related protein, glutamic acid decarboxylase-65, and zinc transport [23-25]. Additionally, B lymphocytes exposed to cell autoantigens produce islet-targeting autoantibodies. CXCR5+ antigen-specific follicular T helper cells promote autoreactive B cells to produce islet autoantibodies. Upon transition into peripheral T helper cells, a subpopulation of follicular T helper cells inhibits the expression CXCR5 and phosphorylates chemokine receptors CCR2, CCR5, and CX3CR1 [17,22]. Immunologic components, which invade the islet of Langerhans and damage insulin-producing cells, produce an inflammatory setting resembling insulitis. This prompts and expedites the establishment of T1DM by enhancing the susceptibility of the immune system to islet antigens carried by human leukocyte antigen class I molecules [26,27]. Immunological control imposed by regulatory T (Treg) cells and programmed cell death protein 1 ligation protects β-cells against autoimmune β-cell apoptosis. Insufficient immunological control can cause autoreactive T cells to initiate an autoimmune attack in T1DM, especially if these cells are activated by β-cells [26]. The destruction of insulinproducing β cells in the islet of Langerhans results in progressive hyperglycemia [28].
Current prevention strategies for T1DM
Several T1DM prevention strategies have been tested in clinical trials (Table 1) [29-41]. Although several studies have found the potential effects of delaying T1DM onset, short effective durations and detrimental toxicities pose challenges in long-term administration. Diabetic pharmacological drugs function by targeting the major signal transduction pathways associated with diabetes pathogenesis. The other pillars of diabetic treatment are nonpharmacological methods including nutritional adjustment, physical activity, and microbiotabased therapy [42]. Both hydrolyzed infant formula and glutenfree diet showed no significant effects in preventing the onset of T1DM [29,30]. According to several immunological studies, early childhood encounter with complex specific proteins may raise the incidence of β-cell autoimmunity in biologically vulnerable individuals [43,44]. Nevertheless, one study found that cow's milk protein plays no role in autoimmunity [29]. Delaying gluten introduction in children also plays no role in the onset of T1DM [30]. The use of vitamins such as vitamin B3, C, D, and E was to no avail [31-33].
The majority of randomized clinical studies examining type 1 diabetes-modifying medication have been undertaken in patients who had been clinically diagnosed with T1DM. Disease-modifying medications such as anti-CD3 monoclonal antibody, rituximab, abatacept, and alefacept substantially maintained insulin release while also exhibiting immunologic impacts [45-47]. Only a few preventive clinical studies are available due to the challenges in sample selection. Patients with T1DM undergo lifetime exogenous insulin replacement treatment as first-line therapy from the moment of diagnosis [48]. Insulin does not prevent or delay the onset of T1DM. Two clinical trials that differed in the assay method for autoantibody detection, micro insulin autoantibody assay versus radioimmunoassay, showed no benefit of oral insulin in preventing T1DM [34,35]. Moreover, the use of alum-formulated glutamate decarboxylase also had no significant effects [36].
TNF-α is a cytokine implicated in the inflammatory process that is generated by activated macrophages, CD4+ lymphocytes, and natural killer cells. TNF-α production may cause loss of pancreatic β cells, leading to the onset of T1DM [49]. Despite the lack of distinction between the groups in the concentration of glycated hemoglobin, the TNF-α inhibitor golimumab promoted improved indigenous insulin levels and decreased the need for insulin therapy when compared to placebo [37]. Moreover, the kinase inhibitor imatinib showed promising results in only the early stages of administration [38]. By addressing insulin resistance and endothelial dysfunction, tyrosine kinase inhibitors exhibit antihyperglycemic properties that have the potential to alleviate or prevent T1DM and type 2 diabetes mellitus [50]. In nonobese diabetic (NOD) mice, type 1 diabetes can be prevented but not reversed with a modest dosage of sorafenib (10 mg/kg/day). However, when sorafenib is administered at high doses (50 mg/kg/day), the drug causes preexisting type 1 diabetes in NOD mice to reemerge. This narrow therapeutic window may be attributed to the inhibition of T helper 1 (Th1) and Tc1 cells in the islets of Langerhans [51]. Similarly, the implementation of the immunosuppressant azathioprine immunosuppressant shows reconstructive insulin production but only in a transient manner [39]. T-celldirected immunosuppression only momentarily prevents the deterioration of cellular functions. The finding that T-celldirected immune intervention results in either no or only temporary retention of cellular functions shows that this therapy does not substantially affect the pathogenesis of T1DM [52]. Other operative approaches such as fecal microbiota transplantation and pancreas transplant alone have proven beneficial in preventing the occurrence of T1DM [40,41]. Nonetheless, there remain concerns, such as the emergence of an instant bloodmediated inflammatory reaction after operative measures, loss of islet volume and density owing to ischemia, islet cell apoptosis, and adverse immunosuppression drug side effects [53].
Dual actions of GABA to delay T1DM onset
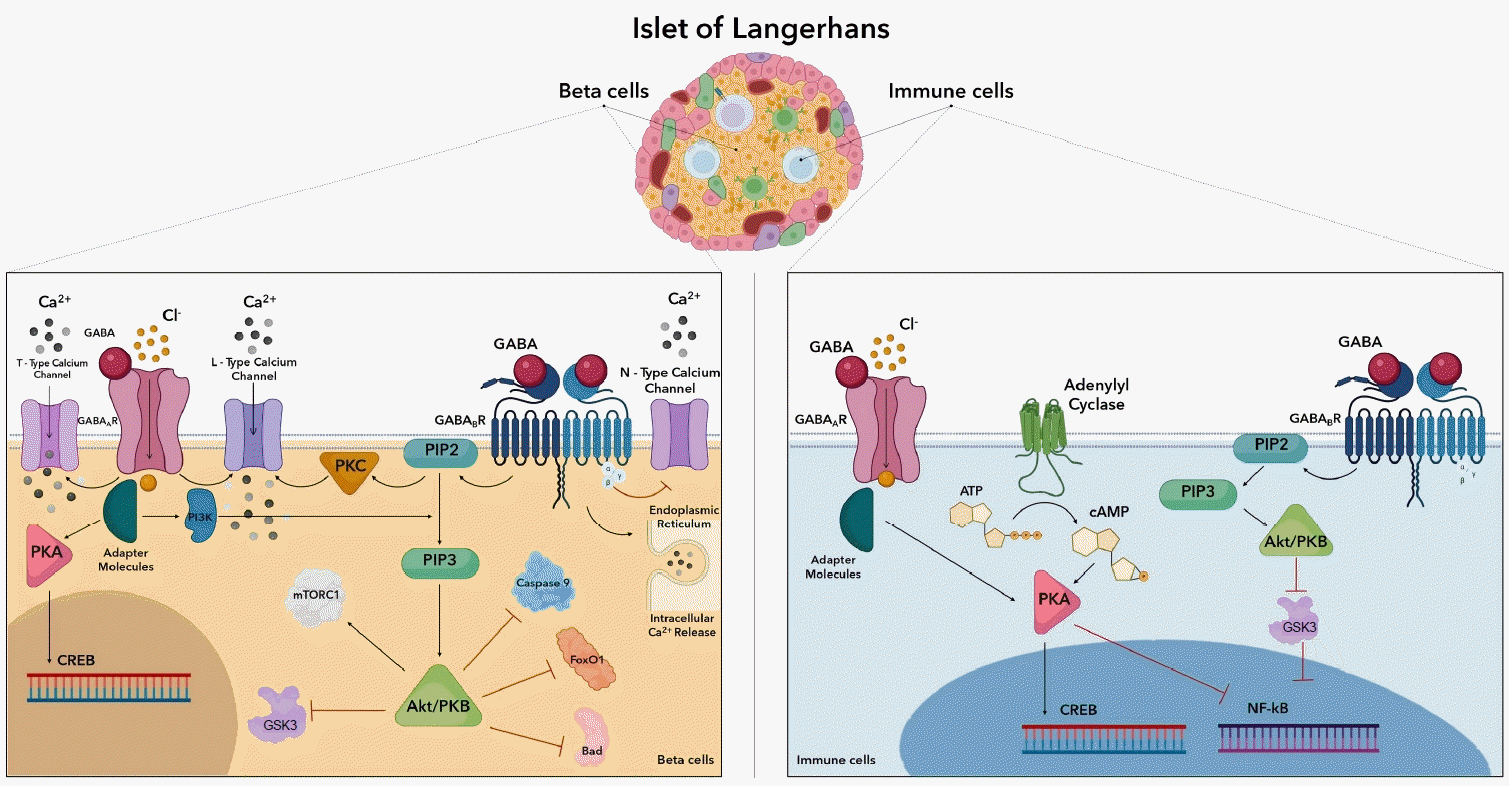
The GABA receptor is a multiunit postsynaptic receptor with 5 components arranged around a central pore. The GABA receptor has 2 classes, GABAA and GABAB, characterized by distinct toxicological, electrophysiological, and biochemical features [54]. GABA plays a significant role in both β cells and immune cells (Fig. 2). GABA receptor-mediated signaling involves adenylyl cyclase, voltage-gated Ca2+ channels, and G protein-activated inwardly rectifying K+ channels [55]. Activation of GABAA receptors leads to an increase in calcium ions through L- and T-type voltage-gated calcium channels. For β-cell GABAB receptors, activation opens potassium channels, releases Ca2+ from intracellular storage, activates protein kinase A (PKA), and stimulates cyclic adenosine monophosphate(cAMP)-response element binding protein (CREB) through a Ca2+-dependent pathway [56]. During maturation, GABAB receptor activation elevates calcium in specific neurons via protein kinase C stimulation [57].
GABAB receptor activation facilitates long-lasting (L-type) current and reduces current through N-type channels, promoting L-type calcium channels current via a non-Gi/o G protein. The Ca2+-dependent signaling pathway triggers phosphoinositide 3-kinase (PI3K)/phosphoinositidedependent kinase (Akt) signaling in response to calcium ion influx [58]. In human β cells, GABA-induced Ca2+ activates the Akt pathway [22]. PI3K catalyzes PIP3 (phosphatidylinositol (3,4,5)-trisphosphate) production, attracting signaling molecules with pleckstrin homology domains, such as protein kinase B and Akt [59]. Induced Akt regulates the phosphorylation of multiple downstream effectors, including glycogen synthase kinase 3 (GSK3), caspase-9, mammalian target of rapamycin complex 1 (mTORC1), Bcl-2-associated death promoter (Bad), and forkhead box protein O 1 (FoxO1) [60]. Akt directly controls cell viability by suppressing pro-apoptotic pathways such as the Bad and FoxO1 transcription factors. FoxO1 proteins either stimulate or repress the transcription of target genes. By modifying the transcription of pancreatic and duodenal homeobox-1 (PDX-1), the PI3K/AKT/FoxO1 signaling pathway can control insulin production [61]. Phosphorylation of Bad also inhibits the apoptotic effects formerly induced [62]. Caspase 9 is a crucial activator of the mitochondrial signaling pathways and a critical pro-apoptotic regulatory protein downstream of the PI3K/Akt pathway. The PI3K/Akt signaling pathway prevents apoptosis by suppressing caspase 9 signal transduction [63]. mTORC1 supports anabolic growth by stimulating protein, nucleotide, and lipid production while inhibiting catabolic mechanisms like autophagy through inhibition of Unc-51-like kinase 1 and transcription factor EB. By stimulation of its downstream targets ribosomal protein S6 kinase beta-1 and eukaryotic translation initiation factor 4E-binding protein 1, mTORC1 stimulates protein synthesis. The PI3K/mTORC1 pathway promotes β-cell development in human islets, and the addition of GABAA caused an increased β-cell area and multiplication [64].
GABAergic stimulation directly affects APCs, reduces MAPK signals, and reduces later adaptive inflammatory reactions to manage active autoimmune disorder [65]. Among T1DM patients, GABA controls the concentration-dependent production of 47 pro- and anti-inflammatory cytokines [66]. GABA may modify the immune system response to pathogens and play a role in autoimmune conditions such multiple sclerosis, type 1 diabetes, and rheumatoid arthritis. Several clinical trials have proven that GABA projects a variety of impacts on the immune system, including activating or suppressing cytokine release, altering cell proliferation, and influencing cell migration [67]. Specific depolarizing channel activation occurs in immune cells as most have ligand-specific gates, such as glutamate receptors and GABAA receptors, which have been found to affect both L- and T-type calcium channels [68]. Adenylate cyclase is a class of enzymes that catalyzes the synthesis of cAMP from adenosine triphosphate and is presumably controlled by a component of the Ca2+-signaling cascade [69]. Similarly, by means of intermediate processes involving intracellular second messengers, a GABAB receptor agonist upregulated basal resting adenylyl cyclase function and facilitated cAMP production. Additionally, cAMP is a significant down-regulator of T-cell function by reducing T-cell immunological activity via the cAMP/PKA/C-terminal Src kinase/lymphocyte-specific protein tyrosine kinase signaling cascade [70]. The transcription factor CREB is generated by PKA. Cell growth, maturation, and survival are all regulated by CREB. CREB regulates T-cell activity and promotes Treg production and stability. Moreover, CREB has been demonstrated to promote transcription of CRE-containing immune-related cytokines such as IL-2, IL-6, IL-10, and TNF-α [70]. Additionally, PKA-dependent activation has the potential to downregulate nuclear factor kappa-lightchain-enhancer of activated B cells (NF-κB) activity. Several studies have proven that increased NF-κB activity is associated with the pathogenesis of autoimmunity, and downregulating this transcription factor would pose therapeutic benefits [71]. The stimulation of GABAB receptor resulted in the suppression of GSK3 expression, which restricted NF-κB activity [72]. Given its numerous roles in cell physiology in T1DM, cAMP has a wide range of modulatory associations on a spectrum of immune cells (Table 2) [66,73-79]. Several studies show that in children who eventually developed T1DM, dysregulation of the GABA metabolism and other circulating metabolites occurred before pancreatic islets autoimmunity [80]. Autoimmunity may therefore be a late reaction to the preceding metabolic abnormalities; thus, adhering to metabolic needs could be an effective delaying or preventive measure.
GABAB receptor agonist in children
Previous studies suggested that by stimulating cell proliferation and survival through the activation of the PI3-K/Akt pathway, GABA is able to delay and reverse the onset of T1D [81,82]. GABA is also able to exert immunoinhibitory effects, precisely by suppressing the activity of Th1 cells against islets, cytotoxic cells; suppressing lymphocyte proliferation and the activation of NF-κB; and increasing the number of Treg cells [83]. GABA is also able to inhibit the secretion of glucagon and has been suggested to have the ability to induce transdifferentiation of α-like cells into β-like cells by acting on the GABAA receptor with the assistance of gephyrin. This results in islet hyperplasia [84]. An experiment showed that GABA-induced differentiation of pancreatic stem cells into insulin-producing islet cells; this was proven by the positive immunostaining results of PDX-1 [85]. PDX-1 and NKx6.1 are 2 of the major markers in β cell lineage investigations [86].
Multiple studies conducted in vivo have shown a potentially positive effect of GABA administration in inducing remission or delaying the onset of T1DM (Table 3). A study in female NOD mice has proven the effect to be dose-dependent as lower doses of lesogaberan (0.025 mg/mL in drinking water) did not produce any significant improvement. The therapeutic effect between doses of 0.25 and 0.75 mg/mL showed little variance [87]. Other studies also identified a dose-dependent relationship between GABA administration and delayed onset of T1DM [88,89]. NOD/SCID mice were given GABA treatment prior to the transfusion of diabetogenic splenic cells. These results show that GABA prevents adoptive transfer of T1DM. GABA also delays T1DM progression in 6-week-old prediabetic NOD mice with autoimmunity. After treatment, at 40 weeks of age, none of the mice were found to be hyperglycemic. The research also suggested that GABA suppresses Th1 activity against islet antigens and consequently inhibits the proliferation of autoreactive T cells [88]. Another study conducted in NSG mice induced with diabetes using streptozotocin showed that oral GABA administration stimulates increase in the mass of β cells, proven by an approximate fivefold increase of β-cell multiplication. The study also concluded that GABA treatment increases insulin levels and exerts protective effects against apoptosis of β cells [56]. Similar conclusions were drawn from another study conducted in NOD/SCID mice in which GABA enhanced islet β-cell proliferation and inhibited β-cell apoptosis [8]. A different study conducted in 8-week-old male FVB mice showed that oral administration of muscimol(GABAA receptor agonist) and baclofen (GABAB receptor agonist) increased proliferation of β cells by 27%±10% and 47%±16% respectively. Baclofen also increased proliferation of α-cells by 72%±21% [90]. Increased expression of PDX-1 and NKx6.1 were found in another study, indicating increased proliferation of β cells. Beta-cell rate of apoptosis also decreased to around 0.5% as compared to 1.5% in untreated mice and GABA and insulin levels increased [86]. A clinical trial proposed to examine the safety and efficacy of GABA in delaying the onset or progression of T1DM in children is currently undergoing phase I clinical trial [91].
Adverse effects and limitations of GABA treatment
The highest concentration of GABA is found in approximately 30% of the brain's neurons and is found in the pancreas at similar levels [92]. GABA is a cerebral neurotransmitter inhibitor which makes CNS depression a frequent adverse effect (AE) [93]. However, oral administration of GABA has been proven to be safe and effective in vivo, oral administration leads to peripheral action and prevents crossing of the blood-brain barrier [83].
The United States Pharmacopeia conducted a review on the safety of GABA. GABA treatment is associated with a brief mild drop (<10%) in blood pressure, abdominal discomfort, headaches, drowsiness, and a brief burning sensation in the throat. All AEs reported ranged from mild to moderate. Multiple clinical studies on GABA use have reported no serious AEs. Importantly, GABA treatment is discouraged in pregnant and breastfeeding women as GABA may cause an increase in growth hormone and prolactin levels. However, no specific studies have mentioned negative effects [94]. That sudden cessation of therapy may lead to withdrawal symptoms is also important to note [95].
Conclusions
Despite substantial advancements in technology, knowledge gaps remain in the understanding and management of T1DM. Multiple preclinical and clinical studies have been conducted to test the potential use of GABA in delaying T1DM, especially in high-risk groups such as children with genetic predisposition for T1DM. GABA exerts dual action on both islet β cells and immune cells. The studies discussed in this article have shown regenerative and protective effects on islet β cells. GABA induces proliferation of β cells and inhibits β-cell apoptosis. No serious AEs related to GABA treatment were reported. These GABA study results create optimism in its potential to delay the onset of T1DM in children.