Highlights
· In conclusion, adrenal hypoplasia congenita (AHC) is diagnosed due to adrenal crisis during the neonatal or infantile period at most of cases. However, it is important to consider adrenocorticotropic hormone when pigmentation or high ACTH are present, even if there is no adrenal insufficiency at an early age.
To the editor,
Adrenal hypoplasia congenita (AHC) is a rare form of primary adrenal insufficiency, typically an X-linked disorder caused by pathogenic variants in the NR0B1 gene, also known as DAX1 [1]. This gene encodes a protein crucial for the development and function of the adrenal glands and the hypothalamus-pituitary-gonadal axis [2]. AHC is characterized by adrenal insufficiency and hypogonadotropic hypogonadism (HH). Adrenal insufficiency often presents in infancy or early childhood with symptoms such as skin pigmentation, hypoglycemia, and salt-wasting dehydration [3].
We report an 8-year-old boy with X-linked AHC caused by a novel likely pathogenic variant in the NR0B1 gene, who initially presented with dark skin pigmentation in the ungual and periungual regions.
The patient had progressive dark pigmentation around the periungual area. He had no siblings and was born at full term with no significant medical or family history of adrenal diseases. His height was 133.5 cm, and weight was 26 kg with in normal range. A physical examination revealed dark pigmentation throughout the body, including the ungual and periungual areas. His testes size was less than 3 mL, but his bone age was advanced to 10 years and 6 months.
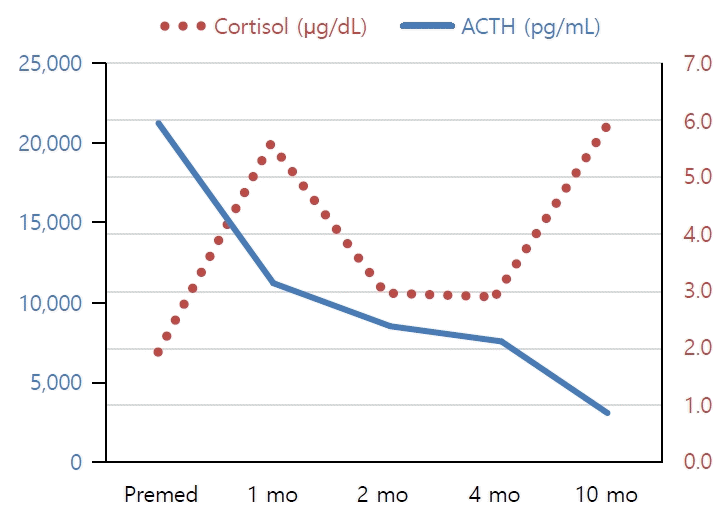
Blood tests indicated no electrolyte imbalance or hypoglycemia, but adrenocorticotropic hormone (ACTH) was elevated (21,310 pg/mL; reference range, 5–37 pg/mL), and cortisol was decreased (2.0 μg/dL; reference range, 4–11 μg/dL), indicating primary adrenal insufficiency. A rapid ACTH stimulation test showed no increase in cortisol or 17-hydroxyprogesterone. Very long-chain fatty acids were not remarkable, and no pathogenic variants were found in the Sanger sequencing of ABCD1 gene. These results suggest that adrenal insufficiency was not caused by adrenoleukodystrophy or 21-hydroxylase deficiency. Abdominal computed tomography showed no abnormalities in the adrenal glands.
Whole-genome sequencing identified a likely pathogenic hemizygous variant, c.134_135del (p.Asp45GlyfsTer26), in the NR0B1 gene, causing a frameshift and premature stop codon [4]. The patient's mother carried the same heterozygous variant but was asymptomatic. As a result, he was diagnosed with X-linked AHC.
NR0B1 is a nuclear transcription factor involved in the steroidogenic pathway in the hypothalamus, pituitary, and gonads [2]. Pathogenic variants in NR0B1 are estimated to affect 1 in 70,000 to 1 in 600,000 males [5]. These variants can lead to primary defects in adrenal gland development and HH, potentially causing absent or delayed puberty and infertility due to spermatogenesis abnormalities [6)].
Clinical features of adrenal insufficiency in AHC vary depending on the location of the NR0B1 pathogenic variants. Approximately 60% of affected males present with adrenal insufficiency in infancy, while 40% have an onset in childhood. Some cases may not show adrenal insufficiency until adulthood, with relatively mild symptoms [7]. Several cases of X-linked AHC have been reported in South Korea, typically presenting with adrenal crises in neonatal or infantile periods (Table 1) [8-13].
Previously reported cases had an onset of adrenal crisis at a neonatal or infantile period; however, the present case had no adrenal crisis until 8 years old but only developed skin pigmentation only. The normal structure of the adrenal gland and the absence of adrenal crisis suggest that this pathogenic variant could result in a partial loss of function for the gene NR0B1.
The disrupted hypothalamic-pituitary-gonadal axis in AHC leads to decreased follicle-stimulating hormone (FSH) and luteinizing hormone secretion, impairing gonadal function, delaying or preventing puberty, and causing infertility [14]. Treatment options include gonadotropin-releasing hormone pulsatile therapy, sequential gonadotropin therapy with FSH and human chorionic gonadotropin, and assisted reproductive technologies like testicular sperm extraction associated with intracytoplasmic sperm injection [15].
In this case, He is now 9 years and 10 months old and receiving an oral hydrocortisone treatment of 11 mg/m2. The serum ACTH levels have decreased gradually (Fig. 1). There is still pigmentation on the hands and feet, but it is improving compared to the first visit. However, he had not yet entered puberty in despite of the advanced bone age; therefore, he must be closely monitored for the onset of puberty.
In conclusion, AHC is diagnosed due to adrenal crisis during the neonatal or infantile period at most of cases. However, it is important to consider AHC when pigmentation or high ACTH are present, even if there is no adrenal insufficiency at an early age. Also, genetic analysis can useful in diagnosing adrenal insufficiency resulting from unknown causes.