Highlights
· Long-read next-generation sequencing (NGS) can provide sequence reads of several kilobases or megabases and therefore identify several pathogenic variants that have been overlooked by other methods.
· Despite its relatively high error rate and high cost, long-read NGS is becoming an important tool for molecular diagnosis of congenital diseases, including endocrine disorders.
Introduction
Nucleotide substitutions and structural variants in the genome play a major role in the development of congenital diseases, including endocrine disorders [1-3]. The identification of pathogenic variants in patients with congenital disorders helps to optimize management procedures and increase the accuracy of genetic counseling [4]. Currently, short-read nextgeneration sequencing (NGS) and array-based comparative genomic hybridization (CGH), together with conventional methods such as karyotyping, Sanger sequencing, and fluorescence in situ hybridization, are used to identify pathogenic mutations and copy number variants (CNVs) [1,2,5]. Still, these methods have several technical limitations. For example, short-read NGS typically yields sequence reads measuring 100 or 150 bp in length and is optimized to detect simple nucleotide substitutions and indels [1]. Likewise, array-based CGH primarily focuses on CNVs in single-copy genomic regions (https://www.agilent.com/en/product/cgh-cgh-snp-microarray-platform). As a result, these methods often overlook chromosomal rearrangements, particularly retrotransposon insertions, repeat expansions, and copy number neutral inversions. These missing genomic variants may account for a certain percentage of cases with congenital disorders. Moreover, short-read NGS and array-based CGH cannot be used to evaluate epigenetic abnormalities.
Recent advances in long-read NGS have enabled researchers to identify several previously unrecognized genetic variants [6-10]. Furthermore, long-read NGS can be used for mutation screening in difficult-to-sequence regions, as well as for DNA-methylation analyses and haplotype phasing [7,9]. Currently, the relatively high error rate and the high cost of long-read NGS are being improved [10]. In this mini-review, we introduce the usefulness of long-read NGS in the molecular diagnosis of congenital disorders, particularly of pediatric endocrine disorders.
Long-read NGS
Nature Methods described long-read NGS as the "Method of the Year 2022. [7]" Long-read NGS is becoming a popular tool for molecular analysis of clinical samples. Two technologies, i.e., single-molecule real-time sequencing by Pacific Biosciences (Menlo Park, CA, USA) and Oxford Nanopore Technologies (ONT) (Oxford, UK), are predominantly used in research and clinical settings [7]. These 2 technologies can provide sequence reads of several kilobases [9] and therefore are capable of characterizing complex structural variants, chromosomal inversions, retrotransposon insertions, and repeat expansions. Of course, long-read NGS can be exploited for the detection of simple nucleotide substitutions and indels, although it leads to a higher error rate in sequence data than short-read NGS. Notably, long-read NGS can be used not only for whole-genome sequencing but also for target sampling for specific regions of interest [8,11]. Target sequencing significantly reduces the cost of sequence analyses. In particular, ONT has provided a system for software-based target assignment designated as adaptive sampling [11]. Furthermore, long-read NGS has advantages in DNA-methylation analysis and rapid workflow [7].
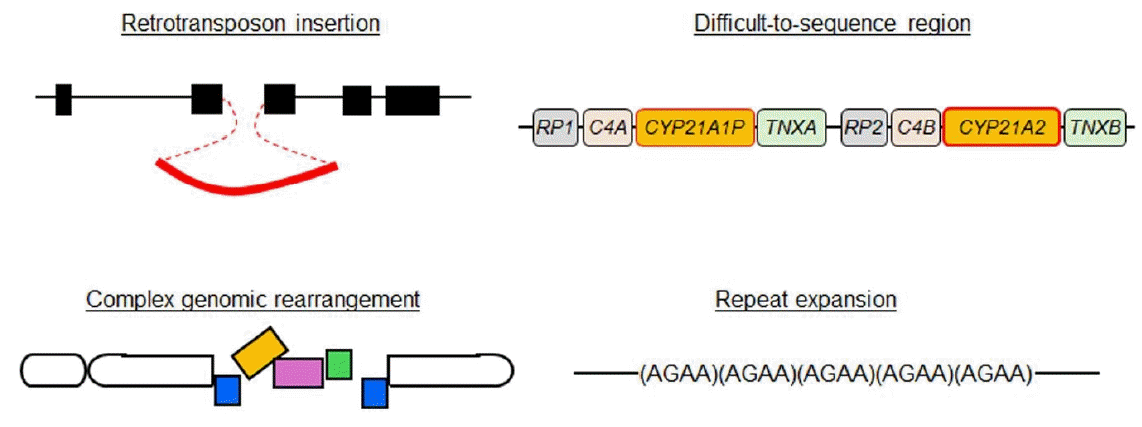
Long-read NGS is particularly beneficial for the detection of specific types of genomic abnormalities (Fig. 1). In the following sections, we provide some examples of molecular diagnoses of endocrine disorders achieved by long-read NGS.
Identification of pathogenic structural variants and repeat expansions
Recently, long-read NGS succeeded in identifying several missing variants associated with congenital diseases [6-10]. Identified variants included CNVs involving transposable elements [6,7,9]. It is known that such elements account for a substantial percentage of the human genome and that insertions of these elements can cause disease phenotypes by disrupting exons or the cis-regulatory machinery of gene expression [11,12]. Nevertheless, CNVs of transposable elements are barely discernible by short-read NGS and array-based CGH. In 2022, Miller et al. [13] performed targeted long-read NGS for a family with autosomal dominant pseudohypoparathyroidism and successfully identified a ~2.8-kb insertion in the GNAS region on chromosome 20. This insertion was a retrotransposon element composing an SVA-VNTR-Alu sequence and is assumed to have caused the disease phenotype through epigenetic dysregulation of nearby genes. Similarly, an Alu insertion was identified in an ALMS1 intron of patients with Alstrom syndrome [14], and an insertion of the SINE-VNTR-Alu retroelement was detected in an NR5A1 intron of patients with disorders of sex development [15]. Many other examples support the usefulness of long-read NGS in the detection of missing structural variants [6,7,9].
Furthermore, long-read NGS is capable of analyzing copy number alterations of repeats and repetitive sequences that are often overlooked by other methods [6,7,9]. Expansions of short tandem repeats, particularly triplet repeat expansions, are known to be one of the major causes of neurological disorders and have also been linked to other types of disorders [16,17]. For example, a trinucleotide repeat expansion in DMD intron was identified in patients with myopathy [18]. Furthermore, Miyatake et al. [19] succeeded in identifying pathogenic repeat expansions in several patients with neurological and neuromuscular diseases. The authors concluded that ONT-based adaptive sampling is superior to conventional diagnostic methods in terms of speed, accuracy, and comprehensiveness.
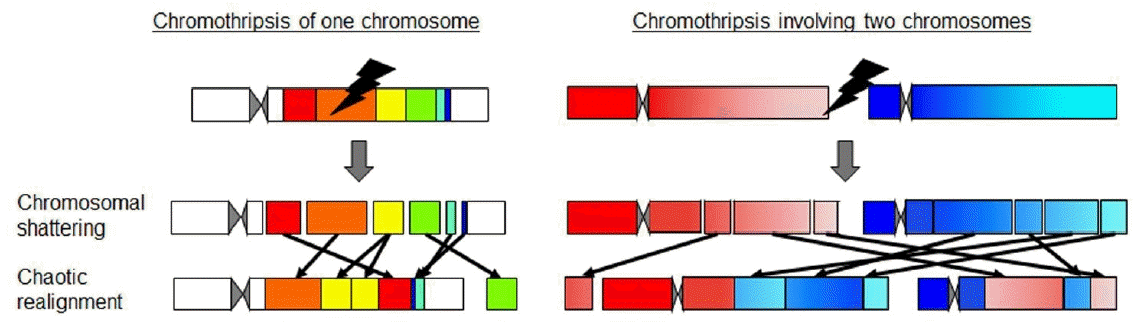
In addition, long-read NGS is beneficial in determining the structure of complex rearrangements [6,7,9]. In this context, recent studies have discovered unique cellular events designated as chromothripsis or chromoanagenesis, which result in highly complex rearrangements involving one or a few chromosomes (Fig. 2) [20-22]. To date, chromothripsis-/chromoanagenesis-compatible rearrangements have been identified in multiple patients with growth failure, congenital malformations, and endocrine abnormalities [20-22]. However, whole-genome sequencing using short-read NGS and arraybased CGH show difficulties in determining the alignments of chromosomal fragments of these cases. Lei et al. [23] employed long-read NGS and determined the structure of a massively rearranged chromosome in a patient with atypical Langer-Giedion syndrome and Cornelia de Lange syndrome type IV. Furthermore, long-read NGS can detect copy number neutral inversions and translocations that are usually undetectable by array-based CGH.
Sequencing for difficult-to-sequence regions
The human genome contains several difficult-to-sequence regions [23]. These regions often harbor homologous sequences due to segmental amplification [23]. An important example of difficult-to-sequence regions from the viewpoint of pediatric endocrinology is 6p21.33, which contains CYP21A2 [24]. CYP21A2 is the causative gene for congenital adrenal hyperplasia due to 21-hydroxylase deficiency [25]. This gene is located within a segmentally duplicated region of approximately 40 kb (Fig. 1) [25]. The presence of a pseudogene (CYP21A1P) renders CYP21A2 difficult for short-read NGS and Sanger sequencing to analyze [25]. Aberrant recombination between CYP21A2 and CYP21A1P is often observed in patients with 21-hydroxylase deficiency [25]. Thus, efforts were made to sequence this region using long-read NGS [26,27]. Adachi et al. enabled mutation screening of CYP21A2 through longread NGS for PCR-amplified DNA fragments [27]. The authors proposed that the cost of this analysis can be reduced by using a barcode system.
DNA-methylation analysis
Long-read NGS can be used to analyze DNA modification [6,7,9]. In particular, ONT-based NGS can analyze DNA methylation without bisulfite treatment of DNA samples. Information on DNA methylation is critical for molecular diagnosis of imprinting disorders, which explain a certain percentage of neonatal diabetes, pseudohypoparathyroidism, pubertal disorders, and short stature in individuals considered small for their gestational age [28]. Yamada et al. [29] performed DNAmethylation analyses using targeted ONT-based NGS and successfully diagnosed patients with Prader-Willi syndrome or Angelman syndrome. Furthermore, ONT-based NGS is useful in the genome-wide screening for DNA-methylation changes associated with disorders (epi-variants).
Haplotype phasing
Another advantage of long-read NGS over short-read NGS and array-based CGH is its capability for haplotype phasing [9]. Haplotype phasing is a method to differentiate 2 alleles inherited from the mother and the father [9]. Haplotype phasing provides critical information on compound heterozygosity of mutations associated with autosomal recessive diseases; this method can determine whether 2 variants in a gene are located on the same or different alleles. Furthermore, it is useful to determine whether a de novo mutation is located on the maternally or paternally derived allele. This information helps to predict the consequences of variants of imprinted genes. The combination of haplotype phasing with DNA-methylation analysis enables researchers to determine epigenetic abnormalities on each allele. In addition, haplotype phasing can be used to clarify the parental origin of each DNA fragment of complex rearrangements and to examine the clonality of multiple mosaic variants in tumor tissues [9].
Limitations and further applications of longread NGS
Long-read NGS has some limitations, which include a relatively high error rate and cost [6,7,9]. However, the accuracy of sequence results is continuously improving, and technical advances are helping to reduce the cost per base [9,10]. It is expected that long-read NGS will become a major platform for molecular diagnosis of congenital disorders. In this context, ONT-based NGS is characterized by rapid workflow [7]. A pilot study at Stanford University showed that ultra-rapid ONT-based NGS was able to diagnose rare genetic diseases of critically ill patients within an average of 8 hours [7]. In addition, long-read NGS has several further applications in research and clinical settings, such as cancer genetics and infection surveillance [7,9], although this is beyond the scope of the present article.