Diverse etiologies, diagnostic approach, and management of primary adrenal insufficiency in pediatric age
Article information

Abstract
Primary adrenal insufficiency (PAI) in pediatric age is a rare, but potentially fatal condition caused by diverse etiologies including biochemical defects of steroid biosynthesis, developmental abnormalities of the adrenal gland, or reduced responsiveness to adrenocorticotropic hormone. Compared to adult PAI, pediatric PAI is more often the result of genetic (monogenic, syndromic disorders) than acquired conditions. During the past decade, rare monogenic disorders associated with PAI have helped unravel the underlying novel molecular genetic mechanism. The diagnosis of adrenal insufficiency in children and young infancy is often challenging, usually based on clinical suspicion and endocrine laboratory findings. Pediatric endocrinologists sometimes encounter therapeutic difficulty in finding the balance between undertreatment and overtreatment, determining how to optimize the dose over the patient’s lifetime, and maximizing mimicry of normal cortisol secretion with glucocorticoid replacement therapy.
Highlights
Primary adrenal insufficiency (PAI) in pediatric age is very rare, but potentially life threatening condition. The diagnosis and management of PAI in children and young infancy is often challenging. The pediatric endocrinologists need to be well aware of diverse causes and management of PAI.
Introduction
Primary adrenal insufficiency (PAI) is endocrinologically defined by the impaired production of glucocorticoids (GC) with or without mineralocorticoid (MC) deficiency, and reduced or excessive adrenal androgens secretion. Plasma adrenocorticotropic hormone (ACTH) is elevated. The most common cause of pediatric PAI is congenital adrenal hyperplasia (CAH) of 21-hydroxylase deficiency (21OHD) [1]. During the past decade, a number of rare monogenic disorders with PAI have been reported. The diagnosis of AI in children and young infants is frequently delayed because it has nonspecific, vague clinical symptoms. Pediatric patients in adrenal crisis is characterized by severe clinical features of hypoglycemia, shock, and coma, leading to life-threatening events. Recently, next-generation sequencing methods have uncovered new genes responsible for causing PAI and enhanced molecular diagnostic yield [2]. Precise endocrine and genetic diagnosis is critical for not only avoiding fatal adrenal crises but also proper genetic counseling to families at risk. The management of pediatric PAI is challenging, in particular optimizing the dose of GC replacement therapy during daily life. This review provides a comprehensive overview of pediatric PAI, focusing on the varying causes of pediatric PAI as well as diagnosis and management.
Diverse causes of pediatric PAI
1. Inborn errors of steroid biosynthesis
CAH is the most common cause of PAI, a group of disorders caused by biochemical defects of steroid biosynthesis inherited in autosomal recessive manner, during infancy and childhood. The most common defect is 21OHD due to mutations in the CYP21A2 gene, accounting for 95% of all types of CAH. The incidence of CAH due to 21OHD detected by newborn screening in the Korean population is 1 in 22,700. CAH of 21OHD is also the most common cause of 46, XX, a disorder of sex development (DSD). The most common mutations in classical Korean forms of the disease are large deletions and the c.293-13A>G, followed by p.I172N and p.R356W [3]. Other types of CAH such as 11β-hydroxylase deficiency, 3β-hydroxylase deficiency, 17α-hydroxylase/17,20-lyase deficiency, congenital lipoid adrenal hyperplasia (CLAH), and P450scc deficiency are less common overall, but interestingly, CLAH is relatively common in Korea.
CLAH is the most severe form of CAH and typically manifests as hyperpigmentation and AI in the neonatal period. CLAH is caused by mutations of the steroidogenic acute regulatory (STAR) gene. The STAR p.Q258* mutation is the most common (88% of allele frequency) in Korean CLAH patients due to founder effect [4,5]. The defect of the CYP11A1 gene, encoding the cholesterol side chain cleavage enzyme P450scc, clinically resembles STAR-related classic CLAH but lacks adrenal enlargement. Nonclassic CLAH (NCLAH) is caused by pathogenic missense mutations in STAR or CYP11A1. Given its overlap with features of familial glucocorticoid deficiency (FGD), NCLAH is sometimes referred to as familial glucocorticoid deficiency type 3 (FGD3) showing ACTH resistance [6]. Most patients with 17α-hydroxylase/17,20-lyase deficiency typically present with hypertension and primary gonadal failure during adolescence and adulthood [7].
Cytochrome P450 oxidoreductase (POR) deficiency is a rare autosomal recessive form of CAH. POR deficiency is caused by mutations in the POR gene, which encodes an electron donor for all microsomal P450 enzymes. The disorder demonstrates a constellation of clinical and endocrine features characteristic of 17α-hydroxylase/17,20-lyase and 21-hydroxylase deficiencies, skeletal dysplasia (Antley-Bixler syndrome), ambiguous genitalia in female newborns, and undervirilization in male newborns [8]. (Table 1)
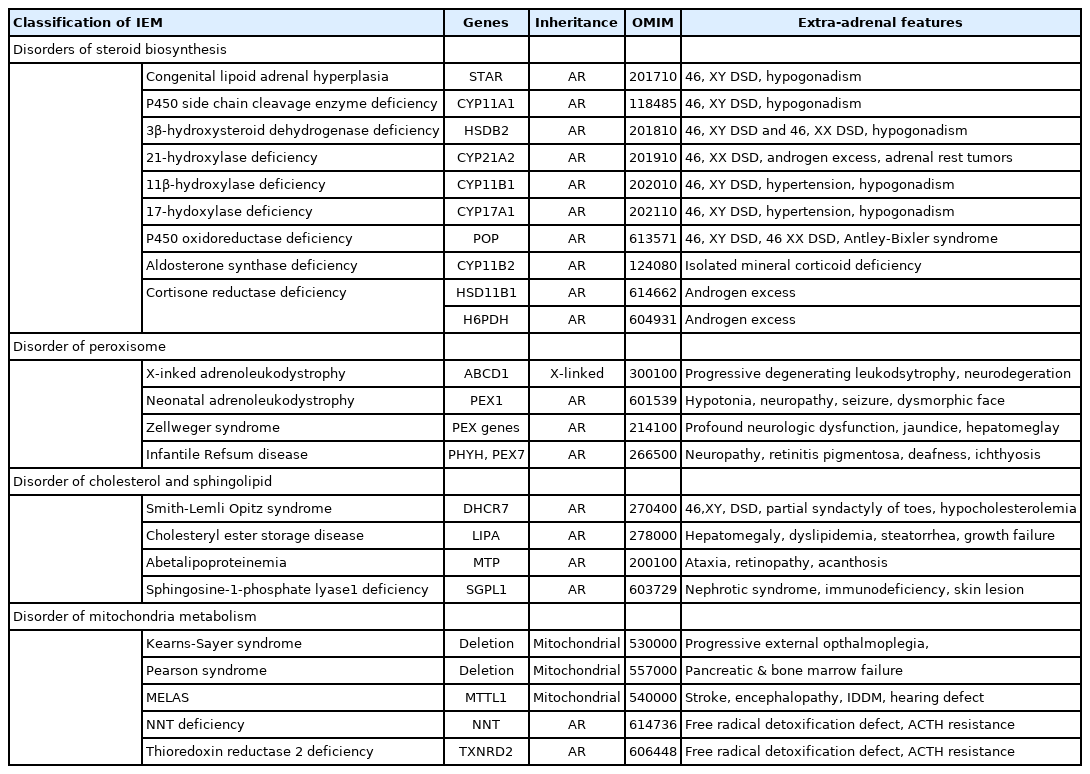
Causes of primary pediatric adrenal insufficiency; inborn errors of metabolism (IEM)
2. Inborn errors of peroxisome biogenesis and enzyme
X-linked adrenoleukodystrophy (X-ALD) is a neurodegenerative disorder associated with PAI due to mutations in the ABCD1 gene, encoding a peroxisomal transmembrane protein. X-ALD is one of the most common causes of pediatric PAI. Affected males are asymptomatic at birth, but can be detected as newborns by tandem mass spectrometry screening. Endocrine and clinical evidence of PAI usually precedes the development of neurological signs in childhood by several years [9].
Zellweger spectrum disorders (ZSD) are extremely rare inborn errors of peroxisome biogenesis, inherited in autosomal recessive fashion, caused by mutations in the PEX genes. They are characterized by liver enlargement, dysmorphic facial appearance, and developmental delay. ZSD range from the most severe phenotype with death in the first year of life (Zellweger syndrome) to attenuated phenotypes (neonatal ALD and infantile Refsum disease). About the one third of ZSD patients have PAI [10]. (Table 1)
3. Inborn errors of cholesterol and sphingolipid metabolism
Smith-Lemli-Opitz syndrome (SLOS or 7-dehydrocholesterol reductase deficiency) is an autosomal recessive disease caused by a DHCR7 gene mutation. Clinical features are developmental delay, dysmorphic features such as Y-shaped partial syndactyly of the second and third toe, and undervirilization in affected males. However, PAI and adrenal crisis are very rare [11].
Cholesteryl ester storage disease (CESD) indicates a spectrum disorder resulting from lysosomal acid lipase deficiency with recessive mutations in the LIPA gene. The most severe form, Wolman disease, presents a rapidly progressive fatal metabolic disorder of newborns with adrenal calcification, jaundice, steatorrhea, vomiting, and failure to thrive. Attenuated phenotypes of CESD present later in life with dyslipidemia, hepatosplenomegaly, and occasional adrenal calcification [12].
Sphingosine-1-phosphate lyase (SPL) deficiency is a new disease causing PAI with other associated disorders such as congenital, steroid resistant nephrotic syndrome, skin lesions, immunodeficiency, and neurological deficits. It is an autosomal recessive disorder caused by mutations of the sphingosine-1-phosphate lyase gene (SPGL1). SPL is an intracellular enzyme catalyzing the final step in the sphingolipid degradative pathway for the removal of sphingolipids [13,14]. (Table 1)
4. Inborn errors of mitochondrial metabolism
Mitochondrial metabolic defects clinically present with dysfunctional symptoms of high energy-requiring organs such as liver, eye, ear, kidney, neuromuscular and endocrine system. PAI is frequently associated with mitochondrial diseases, particularly in syndromic forms of mitochondrial DNA deletions such as Kearns-Sayre syndrome and Pearson syndrome. The other mitochondrial disorders are mitochondrial encephalopathy, lactic acidosis and stroke-like episodes (MELAS) (MT-TL1 on mitochondrial DNA), mitochondrial complex I deficiency (NDUFAF5), and combined respiratory complex deficiencies by QRSL1 mutation [15,16].
There are 2 rare disorders of oxidative stress defects causing isolated PAI: nicotinamide nucleotide transhydrogenase (NNT) and thioredoxin reductase 2 (TNXRD2) defects. Catalase, glutathione peroxidase, and thioredoxin peroxidase enzymes play critical roles in the detoxification of hydrogen peroxide by functioning as antioxidant enzymes. The high concentration of the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) produced by NNT and sufficient TNXRD2 activity are essential for the reduction of oxidized thioredoxin. P450 steroid enzymes are abundant in the adrenal cortex, utilizing NADPH for their catalytic activity, and therefore its function is very sensitive to oxidative stress. TXNRD2 defect also seems to augment the production of reactive oxygen species, aggravating the burden of oxidative stress [17,18]. (Table 1)
5. Monogenic causes of adrenal dysgenesis
Adrenal hypoplasia congenita (AHC) is a rare disorder, mostly inherited in an X-linked pattern. Disruption of the nuclear receptor, DAX-1 (encoded by NR0B1) leads to PAI due to hypoplasia of the adrenal cortex. Patients with AHC usually present with PAI in early infancy with salt-losing phenomena. Hypogonadotropic hypogonadism develops during adolescence. Paradoxically, transient precocious puberty may occur in infancy or early childhood, but eventually these patients end up displaying hypogonadotropic hypogonadism. In less than 10% of AHC patients, deletion of multiple genes located contiguously on chromosome Xp21 cause a contiguous gene syndrome showing the combination of AHC, glycerol kinase deficiency, Duchenne muscular dystrophy, and ornithine transcarbamylase deficiency with intellectual disability [19].
Steroidogenic factor-1 (SF-1/N5A1) is a nuclear receptor that plays a key role in master regulation of adrenal and gonadal development. Heterozygous pathogenic mutations in SF-1/NR5A1 may result in a wide spectrum of DSD. Adrenal function is normal in the vast majority of patients [20].
IMAGe syndrome (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia, and genitourinary anomalies) usually presents with salt-losing PAI in early infancy, caused by a heterozygous gain of function mutation in the cell-cycle repressor gene (CDKN1C) [21]. IMAGe-like syndrome also manifests as PAI, immunodeficiency, and profound postnatal growth failure. It results from autosomal recessive polymerase epsilon-1 (POLE1, Pol +) gene mutations [22]. PAI commonly occurs due to adrenal hypoplasia with variable mineral corticoid deficiency.
MIRAGE syndrome (myelodysplasia, infections, restriction of growth, adrenal hypoplasia, genital phenotypes, and enteropathy) exhibits salt-losing PAI in early infancy. It is caused by a heterozygous gain of function mutation in the growth repressor, the sterile alpha domain containing 9 gene (SAMD9) [23]. The correct diagnosis of syndromic adrenal hypoplasia in PAI patients is challenging owing to its diverse genetic etiologies and overlapping extra-adrenal features. We reported a patient with MIRAGE syndrome who had a SAMD9 mutation and presented with intrauterine growth retardation, AI, and recurrent infection and was initially suspected of having IMAGE syndrome [24]. (Table 2)
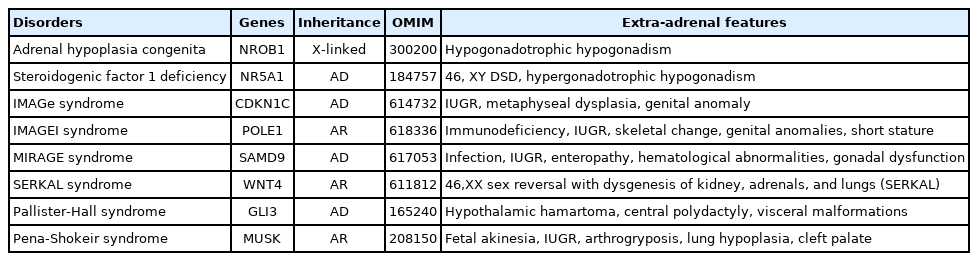
Causes of primary pediatric adrenal insufficiency; monogenic causes of adrenal dysgenesis
6. Monogenic causes of ACTH resistance
FGD is a rare heterogeneous group of PAI characterized by ACTH resistance with decreased GC and mostly normal MC levels. Highly elevated ACTH levels are associated with discernible hyperpigmented skin and mucous membranes. Patients also suffer from failure to thrive, hypoglycemia, and fatigue. FGD1 is most often caused by a defect of the ACTH receptor (melanocortin 2 receptor, encoded by MC2R) [25]. The second most common type, FGD2, results from a defect in the MC2R accessory protein (MRAP, encoded by MRAP), which serves as a cofactor of MC2R to facilitate its trafficking to the plasma membrane [26]. Mild dysfunction of StAR or CYP11A1 activity caused by mild mutations may manifest only as GC deficiency and high ACTH without MC deficiency, or NCLAH [6,27].
Triple A syndrome (AI, alacrima, achalasia of esophagus) results from the disruption of the protein aladin (encoded by AAAS), inherited in autosomal recessive manner [28].
An ultrarare variant of FGD is caused by mutations in the mini chromosome maintenance deficient 4 homolog gene (MCM4), characterized by ACTH resistance, short stature, chromosomal breakage, natural killer cell deficiency, and high risk of cancer and developmental defects [6,29].
Aforementioned oxidative stress defects (NNT and TNXRD2 defects) also cause ACTH resistance syndrome [6,17,18]. (Table 3)

Causes of primary pediatric adrenal insufficiency; monogenic causes of ACTH resistance
7. Monogenic causes of autoimmune disorders
Compared to adults, autoimmune PAI is rare in children. Autoimmune polyglandular syndrome type 1 (APS1) is caused by homozygous mutations in the autoimmune regulator gene (AIRE) and presents with mucocutaneous candidiasis and hypoparathyroidism in early life. PAI and other symptoms such as ectodermal dystrophy usually occur in childhood or later life [30].
IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) is an X-linked recessive disorder representing aggressive autoimmunity, caused by a hemizygous mutation in the FOXP3 gene. It classically manifests in early infancy with enteropathy, autoimmune diseases (particularly type I diabetes mellitus, autoimmune hematologic disorder, nephrotic syndrome), and cutaneous involvement. PAI is rarely associated [31]. (Table 4)

Causes of primary pediatric adrenal insufficiency; monogenic causes of autoimmune disorders
8. Acquired conditions
Acquired causes of PAI are hemorrhage, infiltration, and infection. Symptomatic bilateral hemorrhages are rare but can cause profound adrenocortical dysfunction. Antiphospholipid syndrome (APS) is a thrombotic disorder with antibodies against phospholipids. AI is the most common endocrine problem of APS, originating from adrenal thrombosis and hemorrhage [32].
PAI may develop with the rapid withdrawal of steroids after prolonged therapy. Preterm or sick children under intense stress may have subclinical transient AI, though steroid supplementation is controversial [33]. (Table 5)
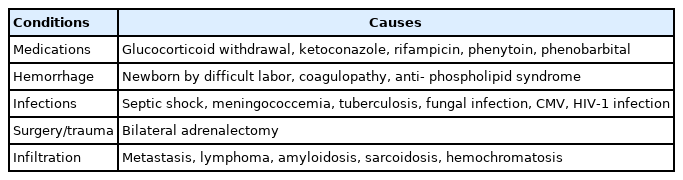
Causes of primary pediatric adrenal insufficiency; acquired conditions
Diagnostic approach
1. Clinical features of PAI and adrenal crisis
Patients with chronic PAI usually suffer from chronic fatigue, failure to gain weight, and nonspecific gastrointestinal symptoms such as anorexia, nausea, vomiting, and recurring abdominal pain. Given that symptoms are vague, mimicking gastrointestinal disease or behavioral and psychiatric problems, it is not easy to make earlier diagnoses. Skin hyperpigmentation may be obvious, particularly in areas unexposed to sun (e.g., palmar creases, areola, axilla, gums), though it is not always clinically evident. Salt cravings are a noticeable symptom of chronic PAI. If not overtly hypotensive, the patient may demonstrate orthostatic hypotension. Adolescent patients also may lose pubic and axillary hair. Patients with acute PAI generally experience altered consciousness, sweating, acute gastrointestinal symptoms, acute dehydration, and shock. Laboratory findings reveal hypoglycemia, hyponatremia, hyperkalemia, and hematologic change. Hypoglycemia is most common in young children. Acute PAI is commonly precipitated by physical and psychological stresses such as infections or surgery and trauma. However, it may also develop without an obvious triggering factor [34,35].
2. Endocrine criteria for the diagnosis of PAI
Endocrine diagnosis of PAI is based on low morning plasma cortisol with an elevated ACTH level, subsequently confirmed by a low stimulated cortisol level. Evaluation for MC deficiency and work-ups for other diseases are crucial. A dehydroepiandrosterone sulfate level below normal for age and sex is a useful initial biomarker of PAI. The diagnosis is highly likely if the basal plasma cortisol level is <140 nmol/L (5 μg/dL) with ACTH concentration elevated more than 2-fold above the upper limit of the reference value for the specific assay method. Unless initial results are definite, a corticotropin stimulation test is needed, which is regarded as the diagnostic gold standard for PAI. Commonly, the standard short corticotropin test using synthetic ACTH analog (cosyntropin) is conducted by evaluating cortisol levels before and 30 or 60 minutes after intravenous or intramuscular bolus injection with a dose of 250 μg for children>2 years of age, 15 μg/kg for infants, and 125 μg for children <2 years of age. A peak cortisol level below 500 nmol/L (18 μg/dL) at 30 or 60 minutes confirms AI. Hyponatremia, hyperkalemia, and elevated plasma renin activity (PRA) strongly suggest MC deficiency [34,35].
3. Stepwise differential diagnosis of diverse PAI etiologies
For the differential diagnosis of PAI etiologies, it is necessary to take into account onset age, phenotypic sex, family history, presence of GC deficiency only or combination of GC & MC deficiencies, presence of genital ambiguity, and extra-adrenal features.
CAH due to 21OHD is the most common cause of AI in infancy with ambiguous genitalia in females, mostly with combined deficiencies of GC & MC, though 25% of cases are late onset and simple virilization type. Neonatal acute onset of AI with GC & MC deficiencies in a phenotypic female without elevation of 17-hydroxyprogesterone (17-OHP) strongly suggests CLAH, particularly in Korea. However, early infantile onset of AI with GC & MC deficiencies in phenotypic males without elevation of 17-OHP strongly suggests AHC, particularly with positive family history indicating an X-linked recessive inheritance pattern. Another type of PAI inherited in X-linked recessive manner is ALD. Elevated very long chain fatty acids is an early biomarker of ALD. PAI with only a GC deficiency may be an initial presenting sign, developing in males between 2 and 10 years of age and usually precedes the onset of neurological symptoms. When a patient’s skin is intensely hyperpigmented, with only GC deficiency and highly elevated ACTH during early infancy and childhood, it strongly indicates a diagnosis of FGD, especially when it recurs in siblings. Notably, there are 8 genes involved in developing endocrine features of FGD (MC2R, MRAP, STAR, MCM4, NNT, TXNRD2, CYP11A1, and SGPL1). Tall stature is also a unique feature of MC2R mutations. Defects of MCM4 and SGPL1 are associated with other extra-adrenal disorders such as natural killer cell deficiency, high cancer risk, and progressive renal dysfunction, respectively. PAI is often the predominant feature of syndromic disorders besides extra-adrenal manifestations. However, it is less often recognized initially because of overwhelming extra-adrenal features. In triple A syndrome, alacrima is often present at birth but is difficult to notice, followed by achalasia and PAI in childhood and adolescence. Newborns with multiple congenital malformations or inborn errors of metabolism may have unrecognized AI. Patients with ZSD have large box-like heads, brain anomalies, and hepatomegaly with prolonged conjugated hyp erbilir ubinemia. IMAGe, IMAGEI, and MIR AGE syndromes share common clinical features such as intrauterine growth retardation, recurrent infection, genital anomalies, and AI. Antley-Bixler syndrome is clinically obvious, with unique craniofacial and skeletal abnormalities. The diagnosis of POR deficiency without skeletal phenotype is problematic since it is characterized by mixed deficiencies of CYP17A1 and CYP21A2, presenting with GC deficiency and mildly increased 17OHP, undervirilized male genitalia, and virilized female external genitalia. Both steroid profiling and genetic testing are helpful to confirm the diagnosis. In early teen-aged, phenotypic female patients with primary amenorrhea and hypertension, CYP17A1 (17α-hydroxylase/17,20-lyase) deficiency is highly suspected regardless of genetic sex. PAI is usually mild with GC responsive hypertension. Patients with Kearns-Sayer syndrome and Pearson syndrome caused by mitochondrial gene deletion often develop PAI with other endocrine dysfunctions such as hypoparathyroidism, growth hormone deficiency, and diabetes, in addition to progressive neuromuscular symptoms. In MELAS, diabetes is more common than PAI. SPL deficiency causes PAI with congenital nephrotic syndrome, skin lesions, and immune deficiency. Adrenal insufficiency is present but often not noted due to steroid therapy for nephrotic syndrome. Among acquired causes of PAI, APS is one of the most difficult etiologies to diagnose. Its nonspecific systemic symptoms, such as prolonged fever and gastrointestinal symptoms, may imitate many other autoimmune or infectious diseases, hampering early diagnosis. Lupus anticoagulant and anticardiolipin antibodies are positive [2,36-38].
Management of PAI
1. Maintenance therapy
Multiple administration is necessary due to the short plasma half-life of hydrocortisone (approximately 90 minutes). In order to mimic the physiologic circadian rhythm, the first and largest dose should be given in the morning after awakening, the second dose after lunch, and the last and smallest dose not later than 4–6 hours before bedtime. A physiologic dose of hydrocortisone for PAI is recommended at 8–10 mg/m2/day in children or 15–25 mg/day in adults, divided into 3 or 4 doses. However, emerging evidence suggests that the dose should be customized because of the wide variability of individual sensitivity to GC, probably caused by polymorphisms in the GC receptor. Notably, alterations in cortisol pharmacodynamics may occur in puberty for many reasons, including changes in 11β-hydroxysteroid dehydrogenase activity, estradiol-related increase in cortisol-binding globulin, and increased glomerular filtration by growth hormone. Altogether, this combination causes reduced circulating cortisol, requiring higher doses. Concomitant medication with CYP3A4 inducers, such as carbamazepine, phenytoin, and barbiturates, results in faster clearance, increasing risk of adrenal crisis by undertreatment. In contrast, inhibitors of CYP3A4, such as erythromycin, lead to increased GC availability [35,39]. The main therapeutic challenge of GC replacement is to maintain the balance between overtreatment and undertreatment. The GC dose may increase according to increasing body surface area. In patients with CAH, GC dosages higher than 20 mg/m2/day in infants and 15 to 17 mg/m2/day in adolescents have been shown to result in loss of final adult height. Thus, meticulous follow-up of growth velocities and general clinical well-being are both important for dose modification. The use of supra-physiological doses of GC for CAH has been associated with obesity, insulin resistance, dyslipidemia and impaired glucose metabolism. The anabolic effect of adrenal androgens may prevent the reduction of bone mineral density (BMD). However, reduced BMD has been reported in relation with cumulative GC exposure during childhood and adolescence [40].
Current GC replacement therapy is not able to precisely simulate the normal physiological circadian rhythm of cortisol secretion. Thus, new strategies and medications for near physiological GC supplementation are under investigation. Oral, modified-release hydrocortisone formulations are promising. Plenadren (Takeda, London, UK) is currently available on the European market for the treatment of adult AI, and provides a rapid increase in cortisol levels after intake in the morning followed by a gradual decrease over the day to almost undetectable levels in the night, similar to the normal circadian rhythm. Chronocort (Diurnal, Cardiff, UK) is another modified-release hydrocortisone under clinical trial that, unlike Plenadren (Takeda, London, UK), can be taken late at night, since it exhibits a delayed and sustained absorption profile for the treatment of adult CAH patients with 21 OHD. A clinical trial of immediate-release granulated hydrocortisone formulation called Infacort (Diurnal, Cardiff, UK) for pediatric use is also underway [41].
Florinef (fludrocortisone) is given in 1 or 2 doses per day at a total of 50–200 μg/day for MC replacement. It normalizes blood pressure, electrolyte balance, and PRA. Newborn infants may require relatively higher doses due to their high resistance to MC. In addition, infants with severe MC deficiency usually require sodium chloride supplementation of 1 to 2 g/day (17 to 34 mEq/day), divided in several feedings, for the first 6 months of life. Children with PAI require a typical daily dose of 100 μg. It is not necessary to adjust the dose of MC by body surface area. The signs and symptoms of inadequate MC replacement include poor weight gain, salt craving, dehydration, hyponatremia with hyperkalemia, and elevated PRA. Excessive MC replacement results in hypertension and suppressed plasma renin.
2. Treatment of acute adrenal crisis
The most important triggering factors include infectious diseases (particularly gastrointestinal infections), perioperative conditions, and exhaustive physical activity. Emergency therapy consists of immediate administration of hydrocortisone as an intravenous 50–100 mg/m2 bolus (higher doses recommended in younger children), followed by continuous infusion or repeated hydrocortisone 50–100 mg/m2/day divided every 6 hours. During less stressful situations (fever, vomiting, minor surgery), the usual doses of oral hydrocortisone are recommended to be doubled or tripled. For the treatment of hypovolemic shock, rapid bolus of normal 0.9% saline 20 mL/kg is indispensable, repeated up to a total of 60 mL/kg within 1 hour. For the treatment of hypoglycemia, 0.5–1 g/kg of dextrose or 2–4 mL/kg of dextrose 25% in water or 5–10 mL/kg of dextrose 10% in water (maximum single dose 25 g) needs to be infused slowly at rate of 2 to 3 mL/min [35].
3. Prevention of acute adrenal crisis
Home management in case of febrile illness includes duplication (>38℃) or triplication (>39℃) of hydrocortisone replacement doses until recovery (usually 2 to 3 days) with the encouragement of profuse electrolyte-containing fluids intake as tolerated. If patients are unable to take oral medication, hydrocortisone is intramuscularly administered 25 mg in infants, 50 mg in children, and 100 mg in adolescents. Minor to moderate surgical stress may require hydrocortisone 50 mg/m2 in children. Otherwise, dose adjustment is not necessary. Major surgery with general anesthesia, trauma, or disease that requires intensive care may necessitate intravenous hydrocortisone 50 mg/m2, followed by hydrocortisone 50–100 mg/m2/day divided every 6 hours with body weight-appropriate continuous intravenous fluids with 5% dextrose and 0.2 or 0.45% saline. Rapid tapering and switch to an oral regimen is possible depending on clinical status. Patients must be educated regarding how to adjust the GC dose during stressful situations and to carry an emergency card [35].
Conclusion
Enhancing awareness of rare genetic causes of PAI is important for pediatricians since the confirmation of a specific diagnosis has implications for therapeutic management and long-term care, including genetic counseling for the family at risk. Furthermore, detailed molecular genetic tests are becoming more available. However, there are still many unmet needs in the diagnosis and treatment of children with PAI because of its diverse clinical presentations and difficulty mimicking physiological changes in cortisol profiles during varying situations. Therefore, cautious clinical evaluation, integrating clinical, endocrinological and genetic findings, is essential for the early diagnosis and appropriate management of children with PAI.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.