Nonclassic congenital lipoid adrenal hyperplasia diagnosed at 17 months in a Korean boy with normal male genitalia: emphasis on pigmentation as a diagnostic clue
Article information

Abstract
Congenital lipoid adrenal hyperplasia (CLAH) is one of the most fatal conditions caused by an abnormality of adrenal and gonadal steroidogenesis. CLAH results from loss-of-function mutations of the steroidogenic acute regulatory (STAR) gene; the disease manifests with electrolyte imbalances and hyperpigmentation in neonates or young infants due to adrenocortical hormone deficiencies, and 46, XY genetic male CLAH patients can be phenotypically female. Meanwhile, some patients with STAR mutations develop hyperpigmentation and mild signs of adrenal insufficiency, such as hypoglycemia, after infancy. These patients are classified as having nonclassic CLAH (NCCLAH) caused by STAR mutations that retain partial activity of STAR. We present the case of a Korean boy with normal genitalia who was diagnosed with NCCLAH. He presented with whole-body hyperpigmentation and electrolyte abnormalities, which were noted at the age of 17 months after an episode of sepsis with peritonitis. The compound heterozygous mutations p.Gly221Ser and c.653C>T in STAR were identified by targeted gene-panel sequencing. Skin hyperpigmentation should be considered an important clue for diagnosing NCCLAH.
Introduction
Congenital lipoid adrenal hyperplasia (CLAH) is a rare autosomal recessive disorder caused by defects in the steroidogenic acute regulatory (STAR) gene located on chromosome 8p11.2 [1]. The true incidence of CLAH is unknown, but the literature suggests it is much higher among Japanese, Koreans, and Palestinians than other ethnic groups. The role of the STAR protein is to transfer cholesterols to the inner mitochondrial membrane from the outer mitochondrial membrane for the synthesis of pregnenolone [2]. Thus, patients with CLAH have deficient levels of glucocorticoids, mineralocorticoids, and sex hormones. Patients with classic CLAH develop adrenal failure within the first few months of life and show female external genitalia regardless of genetic sex [3]. However, some patients with STAR mutations present with mild adrenal insufficiency, even after infancy. These patients are classified as having nonclassic CLAH (NCCLAH), which is caused by STAR mutations that retain partial STAR activity [4].
We present the case of a Korean boy who was diagnosed with NCCLAH at the age of 17 months. During long-term hospitalization for nutritional support necessitated by short-bowel syndrome, whole-body hyperpigmentation and electrolyte abnormalities were noted at 17 months after an episode of sepsis with peritonitis.
Case report
A male child was born at a gestational age of 39 weeks with no history of antenatal or perinatal problems. Apgar scores were 9 points at one minute and 10 points at 5 minutes. His birth weight and height were 3,800 g (z-score, +1 standard deviation score [SDS]) and 52.5 cm (z-score, +1.4 SDS), respectively. There was no family history of consanguineous marriages. The patient was born with an imperforate anus and a colostomy was performed the day after birth. He had normal male genitalia, with both testes palpable, and postaxial polydactyly of the left foot. A grade 2 systolic murmur was heard in the left upper sternal border and echocardiography revealed a patent ductus arteriosus. Bilateral grade 3 vesicoureteral reflux (VUR) was revealed via voiding cystourethrography. Spine ultrasound revealed no vertebral anomalies, and tracheoesophageal fistula was not suspected clinically. Based on this clinical information, including anal atresia, renal anomalies, and limb abnormalities, he was diagnosed with VACTERL association which is defined by the presence of at least 3 of the following congenital malformations: vertebral defects, anal atresia, cardiac defects, tracheo-esophageal fistula, renal anomalies, and limb abnormalities. Laboratory test results, including electrolyte levels, were normal. Neonatal screening tests for metabolic disease were also normal, including the 17-alpha-hydroxyprogesterone level. Chromosome analysis revealed a 46, XY karyotype. He was discharged from the neonatal intensive care unit at 21 days of age. At the age of 3 months, the patient was admitted to the hospital for colostomy takedown, anorectoplasty, and endoscopic dextranomer/hyaluronic acid copolymer injection for VUR. After the surgery, the patient was febrile and hypotensive and his abdomen was distended and discolored. An exploratory laparotomy revealed bowel necrosis. Bowel resection with colostomy and jejunostomy was performed 3 times, which resulted in short-bowel syndrome. He could not be discharged because parenteral nutrition was required. The remnant small bowel extended from the Treitz ligament to the jejunocolonic anastomosis and the length of the remnant bowel was only 40 cm. At the age of 7 months, a takedown of the colostomy and jejunostomy was performed. The patient’s mother observed slight bronzing of the patient’s skin after repeated surgeries
At 17 months of age, the patient had an episode of sepsis and peritonitis secondary to Acinetobacter baumannii infection and an exploratory laparotomy was performed to check for bowel necrosis. At this time, generalized hyperpigmentation was noted by the doctor and the mother stated that the child's hyperpigmentation was markedly exacerbated after exploratory laparotomy for the episode of peritonitis with sepsis.
The patient was referred to our department for the evaluation of the hyperpigmentation. He experienced occasional nausea and vomiting. His heart rate was 120 beats per minute and his blood pressure was 100/65 mmHg. His weight and height were 12.8 kg (1.5 SDS) and 86 cm (2 SDS), respectively. At this point, he was noted to have generalized hyperpigmentation, normal male genitalia, and postaxial polydactyly of the left foot. He was able to ascend stairs with the support of a hand grasp. He could stack 3 cubes, eat without assistance, and pronounce 10 words, meaning that the patient’s developmental status was normal for his age. Laboratory investigations revealed a slightly decreased sodium level (132 mmol/L) but normal potassium (4.8 mEq/L) and glucose (86 mg/dL) levels; complete blood count, blood gas profile, hepatic and renal function, thyroid function test and sex hormone levels were obtained. Decreased cortisol (1.0 μg/dL) and markedly elevated adrenocorticotropic hormone (ACTH) (16,064 pg/mL) levels were noted. The patient's cortisol levels did not respond to stimulation with ACTH (125 μg [250 μg/m2]) (Table 1). Meanwhile, he presented a normal 17-hydroxyprogesterone level (0.42 ng/mL). Plasma renin activity and aldosterone levels were 7.28 ng/mL/hr (normal range, 1.0–6.5 ng/mL/hr) and 0.1 ng/dL (normal range, 3–35 ng/dL), respectively. The abdominal ultrasound revealed no abnormalities, such as adrenal enlargement, hydronephrosis, distal ureter dilatation, or bowel wall thickening. Based on the patient’s symptoms and laboratory results, we considered several genetic causes for primary adrenal insufficiency, and the causative genes of candidate diseases were listed as STAR for NCCLAH and CYP11A1 for congenital adrenal insufficiency. Additionally, melanocortin-2 receptor (MC2R) and MC2R accessory protein for familial glucocorticoid deficiency (FGD) were listed as candidate genes with a low probability because the patient did not have elevated aldosterone.
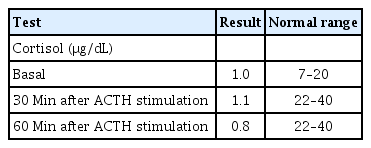
Adrenocorticotropic hormone (ACTH) stimulation test
Targeted gene-panel sequencing was performed to check for pathogenic variants in those genes responsible for primary adrenal insufficiency. Genomic DNA was extracted from the peripheral blood of the patient and both parents. Library preparation was done using the TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA, USA), which enriches a 12-Mb region spanning 62,000 target exons of a total of 4,813 genes. Massively parallel sequencing was performed on the Illumina NextSeq platform. Sequence reads were mapped to the UCSC hg19 standard database for comparative analysis. The average depth of the panel was 85.86X, and percentages of bases above 10X of CYP11A1 and STAR were 99.37%, and 100%, respectively. Meanwhile, no pathogenic variants were detected in CYP11A1 genes.
The exome-sequencing results revealed compound heterozygous mutations for c.653C>T and c.661G>A (p.Gly221Ser) in exon 6 of the STAR gene. Sanger sequencing confirmed the presence of these variants and each of the same heterozygous variant was found in his father and mother, respectively (Fig. 1). This variant has been previously reported in association with CLAH [5-7].
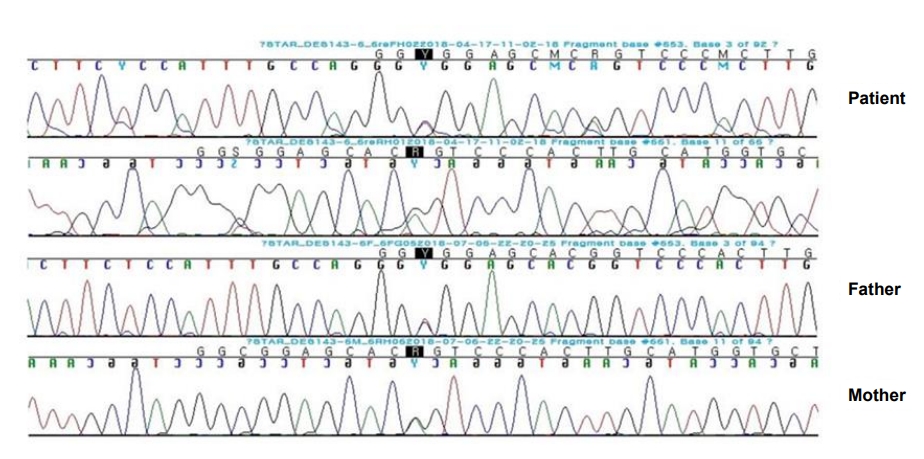
Sanger sequencing confirmation of compound heterozygous mutations for c.653C>T and c.661G>A (p.Gly221Ser) in exon 6 of the STAR gene. Each heterozygous variant was found in the patient’s father and mother, respectively.
Hydrocortisone (7.8 mg/m2/day) and fludrocortisone (50 μg/day) were started as therapy for primary adrenal insufficiency. The dose of hydrocortisone was gradually increased until the age of 24 months for sufficient ACTH suppression and levels of ACTH and cortisol were subsequently decreased to the optimal range (Fig. 2). Finally, at 28 months of age, he was discharged and went home with the attainment of oral feeding without nausea and vomiting. He showed no symptoms of hypoglycemia and continued to show normal growth and development. Our last evaluation of the patient was when he was 36 months of age, during which point, his body weight and length were 16.7 kg (0.5 SDS) and 99.7 cm (1.5 SDS), respectively. Laboratory tests revealed normal levels of cortisol (4.6 μg/dL), sodium (139 mmol/L), potassium (4.9 mEq/L), glucose (70 mg/dL), and ACTH (53.6 pg/mL). The generalized hyperpigmentation had improved, and he continued to visit the outpatient clinic regularly without additional admission.
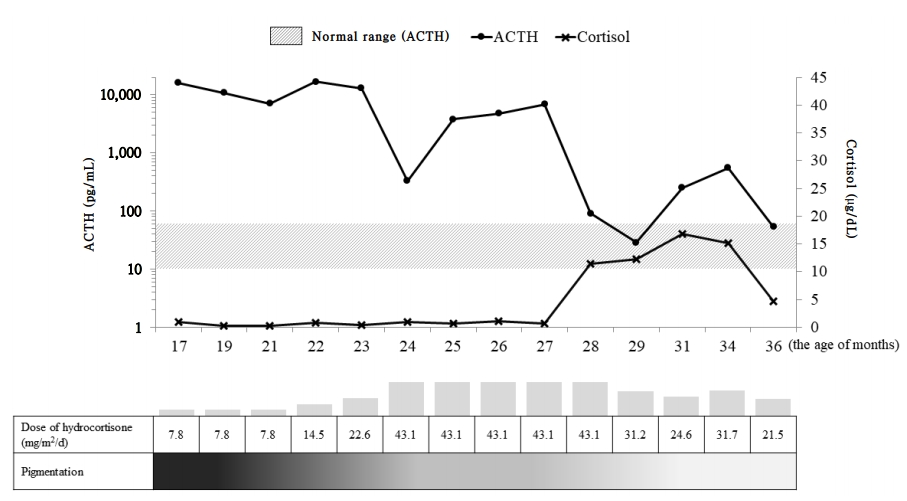
Plasma adrenocorticotropic hormone (ACTH), cortisol, and pigmentation changes during hydrocortisone therapy. ACTH levels are represented by logarithm values.
Discussion
Our patient had generalized hyperpigmentation at 17 months of age and a biochemical investigation revealed a low cortisol level, high ACTH level, and no cortisol response to an ACTH stimulation test. Hyperpigmentation was thought to be a sign of corticotropin hypersecretion due to primary adrenal insufficiency, which can be caused by hereditary steroidogenic enzyme defects, glandular destruction, adrenoleukodystrophy, or congenital adrenal hypoplasia. Both the neonatal screening test and 17-alpha-hydroxyprogesterone levels were normal. Adrenal enlargement, a common finding in congenital adrenal hyperplasia, was not seen on abdominal ultrasound. Other structural abnormalities of the adrenal gland such as hypoplasia, hemorrhage, or tumor were also excluded by abdominal ultrasound. Very-long-chain fatty acids, which are elevated in adrenoleukodystrophy, were normal. Based on the picture of primary adrenal insufficiency with late onset of clinical manifestations, along with the observation of male genitalia and an XY karyotype, we suspected the diagnosis to be NCCLAH, congenital adrenal insufficiency caused by CYP11A1 mutation, or FGD (less likely). The P450 sidechain cleavage enzyme (P450scc), which converts cholesterol to pregnenolone, is encoded by CYP11A1 [8]. Because both P450scc and STAR initiate steroidogenesis, cases of P450scc deficiency have presented with similar features to those of classic CLAH. However, in contrast with classic CLAH, no patient with P450scc deficiency has been reported to exhibit massive adrenal enlargement [8]. The most recent reports of P450scc deficiency included features of NCCLAH with varying degrees of retained adrenal and gonadal function after infancy [9]. Therefore, for individuals presenting with adrenal insufficiency after infancy with normal-sized adrenals, both CYP11A1 and STAR deficiency should be considered; gene sequencing is the most helpful diagnostic method to differentiate between the 2 entities [8].
We performed targeted gene-panel sequencing to screen several genes related to NCCLAH and other types of congenital adrenal insufficiency. No pathogenic variants were found in CYP11A1, while 2 mutations (p.Gly221Ser and c.653C>T) were identified in STAR. Until now, more than 70 different STAR mutations have been described (https://www.ncbi.nlm.nih.gov/clinvar), and 8 mutations have been labeled as causing NCCLAH (Table 2) [6]. In one report, Caucasian siblings (a girl and a boy) with NCCLAH shared the compound heterozygous variants p.Thr44HisfsTer3 and p.Gly221Ser [6]; their ages at diagnosis were 10 months and 14 months, respectively. Relative to our patient, these siblings were additionally hypoglycemic, while the boy similarly had normal male genitalia. The c.653C>T mutation has been reported in 4 patients with classic CLAH who also had p.Gln258Ter in the other STAR allele [5,7]. The c.653C>T variant has been known to lead to skipping of exon 6 or exons 5 and 6 [7]. The p.Gln258Ter mutation is a representative mutation of classic CLAH. One patient with NCCLAH was reported to have the c.653C>T mutation and p.Leu275Pro, which is known as NCCLAH. This patient, with a 46, XY karyotype, presented with symptoms at the age of 2 months and had female external genitalia [2].

Mutations identified in patients with NCCLAH
In vitro functional testing revealed the activity of the p.Gly221Ser and c.653C>T STAR mutations, reported in our patient, to be 30% to 50% and 14% to 24% of that of the wild types, respectively [2,5,6]. Varying clinical manifestations can be seen even in patients with the same mutations [10].
In summary, this report describes a case scenario arising from p.Gly221Ser, c.653C>T STAR mutations in a 17-month-old Korean patient with NCCLAH. Skin hyperpigmentation should be considered as an important clue to diagnosing congenital adrenal disease. We should consider NCCLAH when a patient presents with adrenal insufficiency after the neonatal period or even after infancy.
Notes
Ethical statement
This research was approved by the Institutional Review Board of Samsung Medical Center (approval number: 2012-05-080-009). Informed consent was obtained from the parents of the patient.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.