Childhood obesity and endocrine disrupting chemicals
Article information

Abstract
The prevalence of obesity around the world has increased sharply. Strong evidence has emerged over the last decades that human exposure to numerous endocrine disrupting chemicals (EDCs) is the cause of obesity and obesity-related metabolic diseases. Many EDCs are manmade chemicals that are released into the environment. EDCs are exogenous compounds that interfere with hormonal regulation and normal endocrine systems, thereby affecting the health of animals and humans. The number of chemicals belonging to EDCs is increasing and some of them are very stable; they persist in the environment (persistent organic pollutants). Although they are banned, their concentrations have been continuously increasing over time. This review gives a brief introduction to common EDCs, and evidence of harmful effects of EDCs on obesity-related diseases; we focus in particular on EDCs’ role in causing mitochondrial dysfunction.
Introduction
In modern society, obesity is recognized as a social problem and the World Health Organization (WHO) has called obesity a pandemic spreading worldwide and warned that it is a chronic disease that needs treatment [1]. Over the past 40 years, obesity has also increased rapidly in children and adolescents around the world; in the United States, it has risen to 3 times, with prevalence of 17% in preschool children [2] and in Korea, the prevalence of childhood and adolescent obesity increased from 6.8% in 1998 to 10.0% in 2013 [3]. It is generally accepted that high calorie diet and lack of activity, thus decreased energy expenditure, is a driving force for obesity epidemic. Genetic factors are also considered to play important roles, as the heritability contribution to body mass index (BMI) is known about 25%–60% [4,5].
Then in 2002, Baillie-Hamilton [6] showed evidences that the body's natural weight-control mechanisms are not functioning properly in obesity and the obesity epidemic occurred relatively too quickly, and suggested that environmental causes might be largely responsible. She pointed out that many chemicals have powerful weight-promoting actions. This hypothesis evolved over time and now is known as obesogen hypothesis [6]. In 2011, Neel and Sargis [7] published more accurate data showing close association between prevalence of obesity in the United States and industrial production of chemicals. We added prevalence of diabetes to this picture, which followed exactly the same trend (Fig. 1).
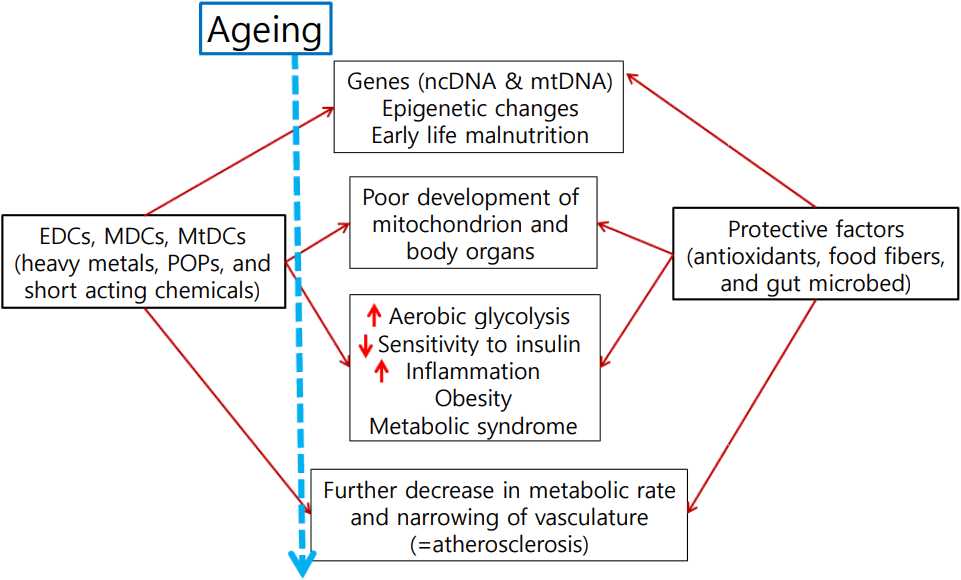
Concepts of the endocrine-disrupting chemical (EDC)-induced mitochondrial dysfunction as cause of obesity. Obesity, insulin resistance and metabolic syndrome are manifestations of poor mitochondrial biogenesis and its function and atherosclerosis is ill-adaptive change (remodeling) of vasculature. ncDNA, nuclear DNA; mtDNA, mitochondrial DNA; MDC, metabolism disrupting chemical; MtDC, mitochondrial function disrupting chemical; POP, persistent organic pollutant. Modified from Lee and Shim. J Diabetes Investig 2013;4:19-33 [49].
Recently Brown et al. [8] published a paper using U.S. National Health and Nutrition Examination Survey data, collected over nearly 4 decades, showing an increase in both caloric intake and BMI over time, but for a given amount of caloric intake, macronutrient intake, or leisure-time physical activity, the predicted BMI was significantly higher in 2006 than in 1998. They concluded that "Factors other than diet and physical activity may be contributing to the increase in BMI over time." There is a consensus, at least among the environmental toxicologists that environmental contaminants are indeed an important cause of the global obesity epidemic [9].
What are these obesogens and how they produce obesity? Before going further into the details, let us examine more general subjects of endocrine disrupting chemicals (EDCs).
Chemicals polluting our environment
Concerns about environmental chemicals began in 1962 after Rachel Carson published the monumental book called "Silent Spring." She claimed that the indiscriminate use of chemicals, especially pesticides, adversely affects the environment. Dichlorodiphenyltrichloroethane (DDT) was developed to prevent malaria epidemic by controlling mosquitoes, but found to cause cancer and other illnesses. The United States Environmental Protection Agency and the United Nations Environment Programme (UNEP) were established in 1970 and 1972, respectively, to pay closer attention to environmental chemicals and human health. United Nations now controls "persistent organic pollutants (POPs)," defined as "chemical substances that persist in the environment, bio-accumulate through the food chain, and pose a risk of adversely affecting human health and the environment," by setting up the Stockholm Convention (http://chm.pops.int/TheConvention/ThePOPs/tabid/673/Default.aspx).
1. Endocrine disrupting chemicals
More than 800 known EDCs are listed in the UNEP-WHO book, and the list is growing. Plasticizers such as phthalates and bisphenol A (BPA) are well known examples. In 2009, the Endocrine Society published the landmark paper "endocrine disrupting chemicals: an Endocrine Society scientific statement" known as "EDC-1" [10], which is followed by a landmark book, "State of the Science of Endocrine Disrupting Chemicals in 2012," by UNEP and the WHO. In 2015, the Endocrine Society published “EDC-2, The Endocrine Society's Second Scientific Statement on Endocrine-Disrupting Chemicals" [11]. This statement described the underlying mechanisms by which exposures during development in animals and humans lay the foundation for diseases later in life. Importantly, it provided a simplified operational working definition of EDCs: an exogenous chemical or mixture of chemicals that interferes with endogenous hormonal axes and induces metabolic disruption in vitro and in vivo. Unlike the early studies of EDCs, which focused on identifying chemicals to modulate the signal transduction of sex steroid and thyroid hormones, emerging data strongly suggest that EDCs disturb the signaling pathways critical for energy homeostasis, mitochondrial activities and insulin signaling.
2. Metabolism disrupting chemicals
Recently environmental toxicologists had started to use a new term, metabolism disrupting chemicals (MDCs), to describe those class of chemicals causing metabolic disturbances [12]. We proposed to use mitochondrial function disrupting chemicals might be better term, as described below. We reasoned mitochondrial function is the key to understanding obesity, insulin resistance, and thus metabolic syndrome, and we used a new term mitochondrial function disrupting chemicals (MtDCs). EDCs could be either MDCs and MtDCs in a book to be published this year. The scheme also is presented in Fig. 1.
3. Mechanisms of actions of EDCs
One of well-known mechanism of actions of EDCs is to alter endocrine function by acting as an agonist or antagonist of the nuclear receptors. Of particular interest is the nuclear receptor family, estrogen receptors (ER), androgen receptors (ARs), thyroid receptors (TRs), and the nonnuclear receptor family, arylhydrocarbon receptor (AhR) [13]. In addition, recent studies have shown that EDCs play a role in regulating pregnane X receptor, the constitutive androstane receptor, and the peroxisome proliferator-activated receptors (PPARs), which act as activators of other nuclear receptors [14]. We will not elaborate on downstream mechanisms after their binding to those receptors, but readers should be reminded that AhR is main receptor binding dioxin like chemicals, thus are frequently called dioxin receptor. Nuclear receptors mentioned above, such as ERs, AhR, and PPARs are also present in mitochondrion and many EDCs are found to move into mitochondrion and disrupt mitochondrial function [15,16].
EDCs as obesogens
1. Molecular pathogenesis of obesity
The molecular toxicological work over environmental compounds that may alter adipose mass is relatively recent, and interest in this area was triggered by initial reports that organotin compounds such as tributylin could increase adipocyte differentiation and adult adipose depots [17]. The effects of tributylin on adipogenesis appear to result in part from its function as a potent PPAR-γ and retinoitc X receptor (RXR) agonist [18,19]. Activation of the RXR/PPAR-γ heterodimer favors the differentiation of preadipocytes and adipocytes progenitors in adipose tissue, and regulates lipid biosynthesis and storage [19]. Organotins, by targeting both RXR and PPAR-γ, thus directly stimulate adipogenesis. Due to this effect, EDCs like tributylin could make increased adipocyte differentiation and adipose tissue amounts in adults following developmental exposure. Triphenyltin has been reported to have hyperglycemic effects and to produce increased circulating insulin concentrations [17].
Following the initial report of the potential role of organotin compounds on adipogenesis, a variety of other chemicals have been reported to have effects on adipogenesis/body weight, glucose tolerance, or diabetes incidence, including BPA, phthalates, and some metals such as arsenic (Table 1). Readers are referred to the recent reviews on this subject [12].
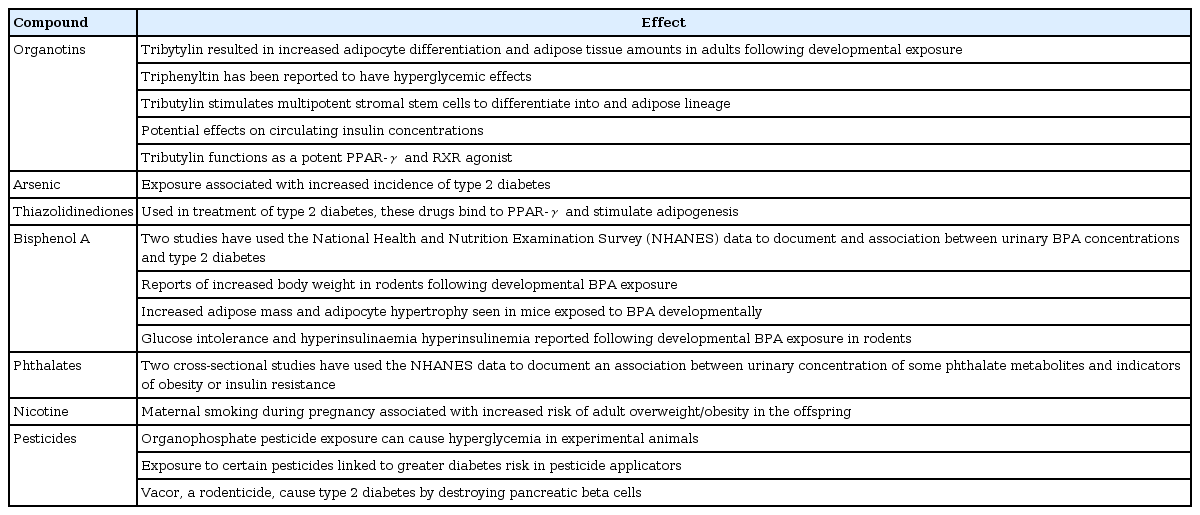
Chemicals with potential effects on adipogenesis, obesity, glucose concentrations, or diabetes
2. EDCs induced mitochondrial dysfunction as the cause of obesity
Mitochondria are one of the intracellular organelles and intracellular power house that produce energy in the form of ATP. Mitochondria have its own genome from its symbiotic origin and is also under the control of the nuclear genome. It has long been known that mitochondrial dysfunction cause insulin secretory dysfunction [20-22]. It is relatively recent that it can also cause resistance [23-25]. We observed that the decrease of mitochondrial DNA density in peripheral blood preceded the type 2 diabetes development in a community-based cohort study, which suggested that mitochondrial dysfunction is the cause of insulin resistance [23]. In that study, the mitochondrial DNA density was also significantly correlated with the components of the metabolic syndrome. The Petersen et al. [24,25] then established the relationship between mitochondrial dysfunction and insulin resistance through a nuclear magnetic resonance spectroscopy and a glucose clamp study. Reduced fatty acid oxidation due to mitochondrial dysfunction results in the accumulation of intracellular fat, increased serine phosphorylation of insulin receptor substrate by increased lipid metabolites, resulting in insulin resistance [26]. Mitochondrial dysfunction involves both genetic factors and environmental factors. Among genetic factors, 16189T>C variant located in the D-loop of mitochondrial DNA in Asian populations is associated with fasting blood glucose and high body mass index, and 1.3 times higher risk of type 2 diabetes [27]. In addition, studies in Koreans and Japanese have reported that the mitochondrial DNA haplogroup is associated with the risk of developing type 2 diabetes [28]. Various environmental factors such as aging and obesity, high fat meals, and environmental disrupting chemicals may also cause mitochondrial dysfunction.
The authors have recently demonstrated that the mitochondrial genome is a thrifty genome and that thrifty traits such as the metabolic syndrome are caused by environmental substances that are toxic to mitochondria. Looking at the US obesity patterns presented by the US Centers for Disease Control over the past decade, we can see 2 surprising facts. The first is that the prevalence of obesity is increasing at a tremendous rate, which is limited to a specific region. The prevalence of the characteristic obesity in the corn belt and its neighboring areas, which are large scale corn cultivation complexes in the United States, may have been caused by environmental factors related to this area.
One possibility is herbicide exposure. Atrazine is the most commonly used formulation in the region and is known to have toxic effects on marine organisms and humans [29]. Atrazine plays an irreversible role in inhibiting electron transport by binding irreversibly to the platoquinone binding site of the photosystem complex II present in the thylakoid membrane in chloroplast [29]. Complex I and III of the mitochondrial electron transport system also have similar Q-binding sites, suggesting that atrazine binds to the mitochondrial Q-binding site and inhibits oxidative phosphorylation of mitochondria.
To demonstrate the hypothesis that insulin resistance and obesity are caused by toxicity to mitochondria caused by environmental toxins such as atrazine, the authors conducted animal studies in which atrazine was given to Sprague-Dawley rats for 5 months [30]. In the atrazine group and the high fat diet group, the body weight was significantly higher, which was associated with mitochondrial abnormalities in skeletal muscle and liver tissue. Atrazine treated group also showed a decrease in basal metabolic rate with increasing fat tissue and increased insulin resistance [30].
3. Biophysical explanation of obesity-metabolic scale law
Kleiber [31] showed that metabolic rate scales with body mass to the three-fourths power. This metabolic scaling law is applicable to elephants, humans, and mice to mitochondria, electron transport system and even molecules [32]. A corollary of this scaling relation shows that unit cellular metabolic power in vivo is negatively correlated with body mass, or that an organism with larger body mass will have smaller unit cellular metabolic power. The metabolic scaling relation is also applicable to within species variations, including human. In other words, when there is a change in body mass, the metabolic rate will change and that change will follow the scaling law.
Why this law is originated is hotly debated. One explanation is that organisms have evolved toward an optimal state in which the energy required for resource distribution is minimized. Another one is body temperature homeostasis; if each cell produce same amount of energy and metabolize same amount of calorie as body size increases, and the heat dissipation does not change, the core body temperature will increase, resulting in burning of organs in the core. In contrast, organisms with a lowered unit cellular metabolic power should gain body weight to compensate for the decreased total heat production and to prevent a decrease in core temperature. This argument is adaptive thermogenesis hypothesis proposed by Dulloo et al. [33] who showed that a low body core temperature precede an obesity in humans. Most of homoeothermic animals including human maintain body temperature at 37℃. As body temperature depends on heat production and divergence and if this balance is broken as in the case of mitochondrial dysfunction induced by EDCs, the core body temperature will fall. The core body temperature regulator can then cause a short-term response such as an increase in the sympathetic nervous system or nonshivering heat production, and piloerection, followed by a long-term reaction, which is the change in body mass and body composition. This long-term adaptation process restores the core body temperature by reducing the heat dissipation through increased body mass and reduces heat dissipation by increased insulating subcutaneous fat tissue, resulting in obesity.
4. Transgenerational effects
Effects of EDCs are not limited to their effects exerted to cells or organisms while they present in the body, but are found in the several generations after the exposure. Experimental animals exposed to environmental contaminants such as BPA, DDT, phthalates, perfluorooctanoic acid, dioxins, and tributyltin during pregnancy are more likely to give birth to offspring that display increased fat accumulation leading to obesity [19,34-40]. The exposure levels used in many of these studies were similar to those measured in human populations. La Merrill et al. [41] showed that exposure to DDT during pregnancy can reduce basal metabolism in the offspring, a fact that could explain why these offspring gain extra weight for a given energy intake. Furthermore, exposure to BPA has been reported to lead to increased food intake due to changes in the brain resulting in stimulated appetite [42]. These transgenerational effects are mediated by epigenetic mechanisms. Authors believe mitochondrial toxicity of EDCs also plays an important role.
Clinical implications of obesogen hypothesis
Obesogen hypothesis demands radical changes in treatment of obese subjects. Most important treatment should be avoidance and elimination of those chemicals, which demand knowledge on the chemicals present in the obese person's body and sources of them. These requirements demand measures of EDCs in human body.
1. How to measure EDCs
Our modern industrial society is awash with man-made chemicals, and we are constantly exposed to innumerable other natural chemicals in food, air, water, and soil. Over 80,000 chemicals are in routine use, and it is estimated that over 1,000 are introduced each year. Over 35,000 pesticides have been registered in the United States, and over 3,000 food additives are approved by the U.S. Food and Drug Administration. Over 700 chemical contaminants have been detected in drinking water [43]. Given the sea of chemicals that we already live in, it is almost impossible to measure the exact quantities of all the chemicals and assess their endocrine-disrupting activities in existing and new chemicals.
Anyhow, POPs are monitored as they are mandated by Stockholm Convention. The standard method is chemical analysis using gas chromatography/mass spectography or high-performance liquid chromatography [44]. This is a highly sensitive test that involves isolation and identifying step for individual contaminants. Since it is not efficient in terms of measurement time and cost, it is difficult to quickly measure a large amount of samples widely spread around us. It requires very expensive analytical equipment and expertise. Furthermore it cannot measure synergistic or antagonistic effects of POPs mixture that occur in vivo, and it is not enough to grasp the toxic effects on the human body in detail. Therefore, a simple, fast and economical high-efficiency bulk search that can replace or complement the present POPs analysis method is required. Much progress has been made in this front, but vastness of the task makes it almost insurmountable.
2. Bioassays
To assess the presence and toxicity of dioxin-like chemicals in environments, toxicologists have been using cell based chemical-activated luciferase gene expression (CALUX) assay. By inserting AhR gene into xenobiotic response element of an appropriate gene, and make it activated when the cell is exposed to dioxin like chemicals, the CALUX assay could be developed as a measure of the toxicity and risks of chemicals at the cellular level [45].
AhR is a ligand-acivated transcription factor belonging to the basic helix-loop-helix/Per-Arnt Sim family [46]. Many toxicological studies have reported that the interaction of AhR ligand with this receptor is involved in a wide variety of toxic and biological effects, such as birth defects, immunotoxicity, neurotoxicity, lethality, tumor promotion, and enzymes (e.g., CYP1A1/2 and CYP1B1) induction [46]. The representative ligand for AhR are dioxins, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), having a very high affinity for this receptor and, therefore, AhR is so-called dioxin-receptor [46]. Environmental chemicals including some pesticides have been identified as AhR agonists, a group that consists of structurally diverse chemicals that are relatively weak inducers of CYP1A1 and/or have markedly lower affinity for AhR than TCDD.
Recently, we developed novel cell-based AhR ligand activity (CALA) assay, a cell line that stably expresses a AhR-responsive luciferase reporter gene construct into mouse hepatoma Hepa1c1c7 cells, without solvent extraction process and analyzed whether low-dose circulating AhR ligands in human serum are associated with parameters of metabolic syndrome and mitochondrial function [15].
3. CALA of serum correlated with serum mitochondrial inhibitor activity on cells
Through this CALA assay, we have shown in a cross-sectional study that the AhR ligand activities were high in glucose intolerance or diabetic patients. In addition, AhR ligand activities were also associated with components of metabolic syndrome such as blood pressure, obesity, and triglycerides. Interestingly, when the serum of the subjects was treated with myoblast cell line, the ability of intracellular mitochondrial ATP production was lower in diabetic and glucose intolerance patients than in normal subjects. In addition, there was a good negative correlation between ATP production capacity and AhR ligand activity. These studies suggest that the AhR ligands in the blood may reduce the mitochondrial function of the tissue and cause diabetes and metabolic syndrome [15].
In a subsequent case-control study that included 83 type 2 diabetic patients, 130 impaired glucose tolerance and 83 normoglycemic subjects, AhR ligand activities were elevated in type 2 diabetes patients and associated with increased type 2 diabetes risk independently of sex, age, and BMI. In addition, in the nondiabetic subjects, AhR ligand activities showed a significant correlation with fasting plasma glucose, fasting insulin concentration, homeostasis model assessment of insulin resistance and negative correlation with adiponectin [47].
We are currently testing if these assays for AhR ligand activities and inhibitory activity of mitochondrial function are predictive of development of T2DM 4 years later with sera obtained from community based cohorts, and to our surprise, the results showed that those with high AhR ligand activity were at increased risk for type 2 diabetes, independently of other diabetes risk factors.
The CALA system thus constructed can be used as a highly sensitive method for detecting dioxin-like chemicals in human body because it allows simple detection of the presence of dioxin like substances in human body (blood) through quantitative measurement by luciferase activity. One of the limitations of this assay is that the individual identification of chemicals is not possible through this test.
Summary
There are strong evidences that EDCs have adverse effects on human health, especially they act as obesogens. We showed EDCs can cause mitochondrial dysfunction, alter cellular metabolic power, and increase body mass. Developmental period is the most sensitive window for EDC action and exposure to this critical period of early development leaves grave consequences to the health of offspring. While it is quite difficult to prove the causality between EDC and obesity, evidences are strong enough to take measures to control human exposure to obesogens in the community [48]. Until then, it is wise to avoid toxicity of POPs by restricting exposure.
Notes
Conflict of interest: No potential conflict of interest relevant to this article was reported.