Congenital hyperinsulinism: current status and future perspectives
Article information
Abstract
The diagnosis and treatment of congenital hyperinsulinism (CHI) have made a remarkable progress over the past 20 years and, currently, it is relatively rare to see patients who are left with severe psychomotor delay. The improvement was made possible by the recent developments in the understanding of the molecular and pathological basis of CHI. Known etiologies include inactivating mutations of the KATP channel genes (ABCC8 and KCNJ11) and HNF4A, HNF1A, HADH, and UCP2 or activating mutations of GLUD1, GCK, and SLC16A1. The understanding of the focal form of KATP channel CHI and its detection by 18F-fluoro-L-DOPA positron emission tomography have revolutionized the management of CHI, and many patients can be cured without postoperative diabetes mellitus. The incidence of the focal form appears to be higher in Asian countries; therefore, the establishment of treatment systems is even more important in this population. In addition to diazoxide or long-term subcutaneous infusion of octreotide or glucagon, long-acting octreotide or lanreotide have also been used successfully until spontaneous remission. Because of these medications, near-total pancreatectomy is less often performed even for the diazoxide-unresponsive diffuse form of CHI. Other promising medications include pasireotide, small-molecule correctors such as sulfonylurea or carbamazepine, GLP1 receptor antagonists, or mammalian target of rapamycin inhibitors. Unsolved questions in this field include the identification of the remaining genes responsible for CHI, the mechanisms leading to transient CHI, and the mechanisms responsible for the spontaneous remission of CHI. This article reviews recent developments and hypothesis regarding these questions.
Introduction
Congenital hyperinsulinism (CHI) is the most common cause of persistent hypoglycemia in infancy, and severe hypoglycemia in infancy can cause permanent brain damage1,2,3,4); therefore, optimal management is extremely important. In the past, our armamentarium against severe CHI was very limited. With only diazoxide and near-total pancreatectomy as available options, many patients were left with psychomotor delay or epilepsy. Even worse, a number of patients developed postoperative insulin-dependent diabetes mellitus3,5,6).
Over the past 20 years, however, remarkable progress has been made in the diagnosis and management of CHI, which has directly translated into improved neurological outcomes for patients3,7). This improvement in the understanding of the pathogenesis of CHI and the development of diagnostic modalities have helped in deciding the optimal management strategy for each patient8,9,10). However, the situation is still far from ideal. Several unsolved questions and unmet needs remain.
In this review, I first discuss the diagnostic criteria and the practical treatment goals for CHI, which are the prerequisites for all subsequent management. Then, after a brief introduction of the mechanism of glucose-induced insulin secretion, I review the current status of the understanding and management of CHI. Finally, I list some of the unsolved questions in this field and introduce key findings that may guide us in the future.
Diagnostic criteria and treatment goals for CHI
1. Diagnosis of CHI (Table 1)

Diagnosis of hyperinsulinemic hypoglycemia
To diagnose hyperinsulinemic hypoglycemia (HI), physicians rely both on clinical clues to identify hyperinsulinism and on laboratory tests to prove hyperinsulinemia.
1) Clinical clues to suspect HI
The presence of HI may be suspected even when the patient is still in an emergency room by asking three questions: When did hypoglycemia develop after the last meal? Does the patient respond to glucagon injection? What is the amount of glucose infusion needed to keep the patient euglycemic?
(1) When did hypoglycemia develop after the last meal?
Euglycemia is maintained by a balance between hepatic glucose output and peripheral uptake induced by insulin. Hepatic glucose output is determined by three factors: food absorption, glycogenolysis, and gluconeogenesis. Fig. 1 shows the duration of glucose production by each of these mechanisms. In disorders of glycogenolysis, the patient typically becomes hypoglycemic after 4-5 hours of fasting. Similarly, in disorders of gluconeogenesis, the patient typically develops hypoglycemia after an overnight fast. When the patient has hyperinsulinemia, hypoglycemic episodes can occur at any time point, sometimes even at 2 hours after the last meal.
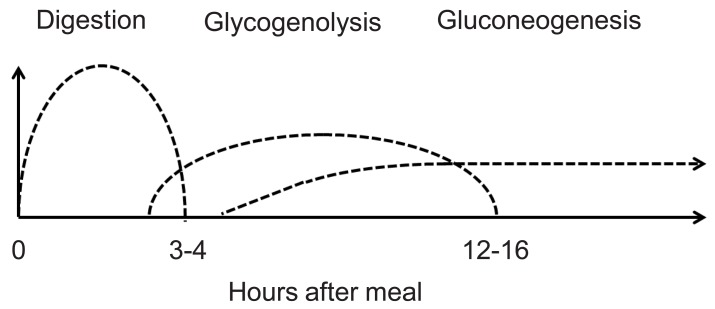
Glucose source during fasting.
(2) Does the patient respond to glucagon injection?
When hypoglycemia is caused by a defect in glycogenolysis, the patient does not respond to intramuscular/intravenous injection of glucagon, which stimulates glycogenolysis. Similarly, when a patient has a defect in gluconeogenesis, by the time the patient becomes hypoglycemic, the glycogen storage in the liver should have been exhausted; therefore the patient does not respond to glucagon either. Only when the patient has hyperinsulinemia can hepatic glycogen be mobilized by glucagon, and glycemic response (>1.7-2.0 mmol/L) is seen.
(3) What is the amount of glucose infusion needed to keep the patient euglycemic?
When hypoglycemia is caused by etiologies other than hyperinsulinism, euglycemia should be maintained by providing the amount of intravenous glucose that corresponds to the normal hepatic (or possibly renal) glucose output: 4-6 mg/kg/min in neonates, 1-2 mg/kg/min in adults, and intermediate values in older children. When euglycemia cannot be maintained by these amounts of continuous glucose infusion, clinicians may suspect the presence of hyperinsulinemia.
2) Laboratory evidence of hyperinsulinism
(1) Insulin at hypoglycemia
HI is diagnosed by demonstrating inappropriately elevated insulin in the presence of hypoglycemia (<2.5 mmol/L, 45 mg/dL). However, it is often difficult to prove hyperinsulinemia by a critical sample taken during a hypoglycemic event11,12). In addition, the term "inappropriately elevated" insulin level is not precisely defined: some authors suggest that any detectable level of insulin is abnormal8,9), whereas others propose different cutoffs13). With regard to the insulin levels during hypoglycemia, "any detectable level" is probably an overstatement because it may suggest but not prove HI. The cutoffs depend on the sensitivity of the particular assay as well as on the insulin sensitivity of each patient. In our own series of 94 confirmed Asian patients with CHI, the insulin at hypoglycemia ranged 8.75-1,250 pmol/L (1.26-180 µU/mL) with a median of 73.3 pmol/L (10.55 µU/mL; unpublished data). In contrast, insulin levels during hypoglycemic events in patients without HI ranged from undetectable to 43.1 pmol/L (6.2 µU/mL) while the detection limit was >2.1 pmol/L (0.3 µU/mL). Clearly, these values overlap.
(2) Relatively low free fatty acid and ketone bodies at hypoglycemia
Insulin inhibits lipolysis; therefore, low free fatty acid and ketone bodies during hypoglycemia are also used as diagnostic adjuncts. In normal infants (0-24 months of age), blood 3-hydroxybutylate and free fatty acid levels after a 20-hour fast are 3.11 mmol/L (range, 1.29-4.34 mmol/L) and 2.15 mmol/L (range, 1.03-3.24 mmol/L), respectively14). These values can be used to set cutoffs for HI. In our series of 207 cases with confirmed CHI, the highest 3-hydroxybutylate at hypoglycemia was 0.44 mmol/L (unpublished data).
3) Proposed diagnostic criteria to suspect HI
It is difficult to set definitive diagnostic criteria for HI. Several authors propose different cutoff values to diagnose HI (Table 1), but these criteria should be regarded as suggestive and not necessarily diagnostic. The bottom of Table 1 includes proposed criteria that strongly suggest the presence of HI in children.
2. Treatment goals of CHI
The goals of HI treatment are to prevent neurological sequelae of hypoglycemia. Factors that could affect neurological outcomes include age, comorbid conditions, severity of the initial episode, and duration and frequency of subsequent hypoglycemic episodes15,16,17,18). Therefore, the treatment goals should be individualized. Currently, blood glucose >3.33-3.89 mmol/L (60-70 mg/dL) is the most commonly recommended target for HI treatment10,11,13).
Insulin secretion and the ATP-sensitive potassium channel
1. Glucose induced insulin secretion (the GSIS pathway) (Fig. 2)
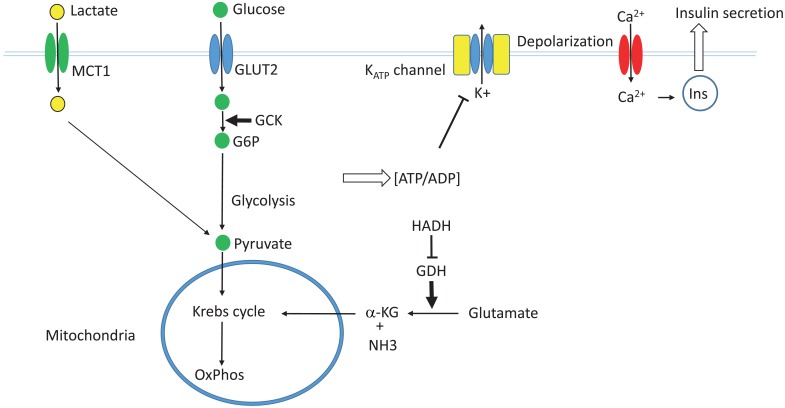
The glucose-induced insulin secretion pathway. GLUT2, glucose transporter 2; GCK, glucokinase; G6P, glucose 6-phosphate; MCT1, monocarboxylate transporter 1; GDH, glutamate dehydrogenase; HADH, L-3-hydroxyacyl-coenzyme A dehydrogenase; α-KG, α-ketoglutarate; Ins, insulin.
In pancreatic β-cells, extracellular glucose is transported into the cytoplasm by the action of glucose transporter (GLUT2). The glucose is then phosphorylated by glucokinase. Glucokinase is not easily saturated by the physiological range of intracellular glucose, and is not inhibited by its end product glucose-6-phosphate. Therefore, it serves as the fuel gauge of the β-cells. Glucose-6-phosphate is then metabolized via glycolysis, the Krebs cycle, and oxidative phosphorylation to generate ATP. An increased ATP/ADP ratio within β-cells leads to the closure of the ATP-sensitive potassium channel (KATP channel), which causes depolarization of the cell membrane and opening of the voltage-gated calcium channel. The resulting influx of calcium ions then causes fusion of the insulin secretory granules with the cell membrane and secretion of insulin19).
2. The KATP channel
The KATP channel is an octameric structure composed of four molecules of pore-forming Kir6.2 and four molecules of SUR1 that surround the pore and regulate the channel activity. Intracellular ATP binds to Kir6.2 molecules to inhibit the channel activity, whereas MgADP binds to SUR1 to activate the channel. Therefore, channel activity is controlled by the ATP/ADP ratio within the cells.
In the endoplasmic reticulum, Kir6.2 and SUR1 associate with each other to form the channel that is transferred to the Golgi apparatus and to the cell surface. If Kir6.2 and SUR1 do not associate with each other, they cannot escape the endoplasmic reticulum and are degraded there.
Kir6.2 and SUR1 are encoded by KCNJ11 (1 exon) and ABCC8 (39 exons), respectively. These genes are located side-by-side with close proximity on 11p15.1.
Known etiologies of CHI (Table 2)

Genetic causes of congenital hyperinsulinism
1. Transient and persistent CHI
There are two main types of CHI: transient CHI, which usually develops soon after birth and resolves spontaneously within the first 3-4 weeks of life and persistent CHI, which can develop later in life as well as in the neonatel period, and lasts longer. The distinction between transient and persistent CHI is not possible on the basis of laboratory test results. In our national survey in Japan, only shorter gestational age and lighter birth weight were predictors of transient CHI20). The incidence of persistent CHI is generally estimated as 1 in 50,000 live births9) although the incidence could be higher in certain populations (e.g., 1 in 2,500 births in Saudi Arabia). On the contrary, the incidence of transient CHI is much higher. In the national survey in Japan, the incidence of transient CHI (1 in 17,000 births) was approximately twice as high as that for persistent CHI (1 in 35,400 births)20).
1) Transient CHI
Transient CHI is believed to be caused mainly by nongenetic factors, e.g., small size for the infant's gestational age or stressful perinatal conditions such as cardiopulmonary disorders. An important exception is the monoallelic inactivating mutation in HNF4A21,22,23). Unlike other patients with transient CHI, patients with HNF4A mutations are often born large for gestational age. Importantly, a fraction of these patients develop a form of dominantly inherited diabetes, maturity-onset diabetes of the young type 1 (MODY1), later in life and therefore should be followed up after resolution of CHI (21-23). Because HNF1A is in the same pathway with HNF4A and its mutation is the cause of MODY3, researchers checked for mutations in HNF1A in patients with transient CHI, and indeed found some patients with mutations in HNF1A24,25).
2) Persistent CHI
In contrast, persistent CHI is believed to have genetic etiologies. However, even with the most comprehensive analysis, the responsible genes can be identified in only 53% of diazoxide-responsive CHI patients26) although in unresponsive patients, KATP channel mutations could be identified in most (87.6%-88%) cases26,27).
2. Causes of persistent CHI
Table 2 lists known causes of CHI. The most common of these are inactivating mutations in one of the KATP channel genes, ABCC8 or KCNJ11 (KATP-CHI). The second most common is an activating mutation of glutamate dehydrogenase (GDH-CHI). Others are relatively rare. When confined to families with consanguinity, inactivating mutations in L-3-hydroxyacyl-coenzyme A dehydrogenase (HADH-CHI) are the most common cause26,27).
1) KATP-CHI
Three distinct subtypes of KATP-CHI are known:
(1) Recessively-inherited KATP-CHI
Recessive KATP-CHI is caused by biallelic mutations in one of the KATP channel genes. This is the most severe form of KATP-CHI, and all β-cells in the pancreas present in abnormal (diffuse) form. Pathologically, recessive KATP-CHI is characterized by large β-cells with abnormally enlarged nuclei28).
(2) Dominantly inherited KATP-CHI
Dominant KATP-CHI is caused by a monoallelic mutation in the KATP channel genes. The presentation is usually relatively milder, and patients often respond to diazoxide29) although there are some refractory cases30).
(3) Focal KATP-CHI
i) Pathogenesis
In patients with focal KATP-CHI, abnormal β-cells are confined to a restricted region in the pancreas. In close proximity with the KATP channel genes at chromosome 11p15.1, an imprinted region at 11p15.5 harbors maternally expressed tumor suppressors, H19 and CDKN1c, and a paternally expressed growth factor gene, IGF2. The focal lesion arises in an individual with a paternally inherited, monoallelic mutation in one of the KATP channel genes. When segmental paternal uniparental disomy occurs as a somatic mutation during the development of the pancreas, that particular cell loses KATP channel activity. At the same time, the tumor-suppressor activities of H19 and CDKN1C are lost, and the activity of IGF2 is doubled. This leads to a growth advantage for the abnormal β-cells and eventually leads to formation of a focal lesion31,32,33,34). Histologically, the focal lesion is characterized by the presence of large β-cells with enlarged nuclei similar to those of the diffuse lesion, and β-cells outside the focus have normal histology35,36,37).
ii) Clinical implication
Although 96.2% of focal lesions are unresponsive to diazoxide3), when the focal lesion is identified preoperatively, partial pancreatectomy can cure the patient without postoperative complications. Therefore, the identification and localization of focal lesions are extremely important. However, because they are generated during the normal organogenesis of the pancreas, they cannot usually be detected using conventional imaging modalities such as computed tomography (CT), magnetic resonance imaging, and angiography. The focal lesions can be preoperatively identified using molecular analysis and 18F-fluoro-L-DOPA positron emission tomography (18F-DOPA PET) scans, thereby enabling surgeons to plan the surgical procedure and to find the lesion intraoperatively.
iii) 18F-DOPA PET scan
18F-DOPA is incorporated into β-cells by DOPA-decarboxylase, which is abundant in β-cells. Following the initial description of its role in identifying the focal lesion38), its usefulness has been reported in a number of publications39,40). 18F-DOPA PET detects focal lesions as small as 5 mm and is better preformed as PET-CT. However, there are some challenges in interpreting the results. First, artifact uptakes tend to be found in the head of the pancreas because the head has a larger mass than the rest of the pancreas and because 18F-DOPA is excreted into the bile duct. Second, 18F-DOPA PET does not necessarily show the exact size of the lesion, especially when the lesion extends so-called tentacles out of the central lesion. These problems appeared more pronounced in our experience in Japan41).
iv) Epidemiology of focal KATP-CHI
Previously, it was reported that approximately 40%-60% of surgically treated patients had focal CHI31,42,43). However, recent molecular analysis has revealed a racial disparity in the frequency of paternally inherited monoallelic mutation in KATP-CHI patients. For example, the percentage of patients with a paternally inherited monoallelic KATP-channel mutation is 20.6% in Spain44), 33% in Norway45), 84.2% in Japan46), 37.7% in Germany47), 58% in China48), and 25% in the UK27). Obviously, these figures could be affected by ascertainment biases and by small sample sizes. In Japan, we have presently identified 46 patients with KATP-CHI, and 37 (80.4%) have paternal mutations. Therefore, combined with the report from China48), it appears that Asians tend to have a higher frequency of paternally inherited mutation, suggesting the presence of focal CHI. The identification of focal CHI, therefore, is even more important for Asian patients.
v) Discordant molecular and 18F-DOPA PET results
Not all patients with a paternally inherited KATP channel mutation have focal uptake by 18F-DOPA PET, and some of these actually show diffuse histology. For example, Banerjee et al.49) reported that 31% of patients with a paternal monoallelic mutation showed diffuse uptake on 18F-DOPA PET. Similarly, Kapoor et al.27) reported that 26% of patients with paternal mutation appeared to have a diffuse lesion on PET. These results may indicate that not all maternal mutations were identified by the molecular analysis. However, when monoallelic mutations were identified in patients with KATP-CHI, the majority were paternal mutations: 79.3% and 65% in two UK series27,49), 84.2% in Japan46), and 83.3% in Norway45). Selectively missing the maternal allele during the molecular analysis is statistically highly unlikely. Another possibility, therefore, is that these patients have unusually scattered focal CHI that resembles true diffuse CHI. Further analysis is necessary to address this problem.
2) Non-KATP channel CHI
Most other persistent CHI are caused by excessive anaplerosis (replenishment of metabolic intermediate) into the GSIS pathway. With the exception of GCK and SLC16A1 mutations, these non-KATP-channel CHI are usually responsive to diazoxide.
(1) GDH
GDH is encoded by GLUD1 at chromosome 10q23.3. GDH mediates conversion of glutamate to α-ketoglutarate and ammonia, which is one of the major anaplerotic pathways in the Krebs cycle. An activating mutation in the GLUD1 gene then supplies excess α-ketoglutarate into the Krebs cycle. The resulting overproduction of ATP causes CHI associated with hyperammonemia (hyperinsulinism-hyperammonemia syndrome)50). Both dominantly inherited and sporadic cases have been reported. Hypoglycemia is responsive to diazoxide, but hyperammonemia is resistant to conventional treatment. Because GDH is allosterically activated by leucine, GDH-CHI presents with the typical leucine-sensitive CHI.
3) HADH
HADH-previously known as short-chain hydroxyacyl CoA dehydrogenase-is encoded by HADH at 4q22-q26. HADH functions in the mitochondrial matrix to catalyze the oxidation of straight-chain 3-hydroxyacyl-CoAs as part of the β-oxidation pathway. Unlike other proteins in the β-oxidation pathway, HADH is abundant in pancreatic β-cells and inhibits the activity of GDH. Biallelic HADH mutation then causes activation of GDH and hyperinsulinemia51,52,53).
4) Glucokinase
Glucokinase is encoded by GCK at chromosome 7p15. Patients with GCK-HI have an activating mutation in GCK. This leads to overactivity in the GSIS pathway and oversecretion of insulin54,55,56). On the contrary, inactivating monoallelic mutation is a cause of MODY2 or GCK-MODY57). Recently, a somatic activating mutation in GCK has been proposed as a cause of a novel form of diazoxide-responsive focal CHI58).
5) Uncoupling protein 2
Mitochondrial uncoupling protein 2 (UCP2) is encoded by UCP2 at chromosome 11q13. UCP2 protein leaks protons across the inner mitochondrial membrane, thereby uncoupling the oxidative phosphorylation from ATP generation. Patients with a monoallelic mutation in UCP2 have excessive ATP production leading to HI59).
6) Monocarboxylate transporter 1
Monocarboxylate transporter 1 (MCT1) encoded by SLC16A1 at 1p12 is in the cell membrane and transports extracellular lactate and pyruvate into the cells. In pancreatic β-cells, the activity of MCT1 is normally suppressed to prevent lactate influx during exercise. In patients with exercise-induced HI, researchers identified mutations in the promoter of SLC16A1 that activate the transporter. During exercise, extracellular lactate is fluxed into the β-cells and is converted to pyruvate, which fuels the GSIS pathway. The resulting increase in ATP production leads to HI60).
3. Syndromic CHI
A variety of syndromes are reportedly associated with CHI. Because CHI is not a common feature of these syndromes, some of these associations may be coincidental. Nevertheless, CHI is frequently associated with Beckwith-Wiedemann syndrome, Sotos syndrome, Kabuki syndrome, Costello syndrome, mosaic Turner syndrome, or congenital deficiency of glycosylation61). In these syndromes, the association may have biological implications more than by-chance association. Of note is the Usher-CHI syndrome62,63) in which CHI is associated with the symptoms of Usher syndrome, i.e., hearing loss and retinitis pigmentosa. This association is caused by biallelic deletions encompassing the KATP channel genes at 15p11 and the adjacent USH1C gene at 11p14.3, which is responsible for Usher syndrome.
Current treatment strategies
Current treatment strategies are summarized in Table 3 and are reviewed below.

Treatment for congenital hyperinsulinism
1. Diazoxide
Diazoxide is a benzothiazine derivative that acts on the SUR1 subunit of the KATP channel, activating it. Diazoxide is used orally in three divided doses (5-15 mg/kg/day) and is effective for a variety of CHI subtypes64). However, it is generally ineffective for the most severe, neonatal-onset, recessive, and focal forms of KATP-CHI. Dominant KATP channel CHI often responds to diazoxide, although some unresponsive cases have been reported32). Hypertrichosis occurs in most patients and could be a serious concern. Other side effects include water retention, which could cause serious problems such as congestive heart failure or reopening of the ductus arteriosus65,66). This side effect may be of particular concern in patients with a low birth weight, as in transient CHI. Routine coadministration of diuretics is advised.
2. Octreotide
Octreotide is a somatostatin analog that acts on the somatostatin receptors SSTR2 and SSTR5 and inhibits secretion of a variety of hormones, including gastrin, cholecystokinin, glucagon, growth hormone, secretin, pancreatic polypeptide, thyroid stimulating hormone (TSH) vasoactive intestinal peptide, and insulin. Although its use for CHI has not been licensed in any country, it has been used for nearly 20 years for both short- and long-term control of diazoxide-unresponsive CHI67,68). It is administered as multiple daily subcutaneous injections (3-4 times/day) or by continuous subcutaneous infusions using an insulin pump. In our experience, many patients with KATP channel CHI can be maintained on long-term treatment until spontaneous remission at 2-5 years of age69). Common adverse events include gastrointestinal symptoms, white stool, dilated gall bladder with or without gall stones, and growth deceleration after 2 years of age. Rarer, but more serious side effects, include hepatitis70), necrotizing enterocolitis71) and long QT syndrome72).
3. Glucagon
Glucagon stimulates glycogenolysis and gluconeogenesis to increase hepatic glucose output. It is administered by intravenous, subcutaneous, or intramuscular routes, and has been used mainly for short-term control of diazoxide-unresponsive patients who are not adequately controlled by other means. However, as is the case for octreotide, its long-term use until spontaneous remission has been reported73,74). Apart from its gastrointestinal side effects, its crystallization in the infusion tubes has been a major practical problem during long-term use. Part of this problem may be ameliorated by the development of a water-soluble formulation that is in a phase 2 clinical trial (ClinicalTrials.gov Identifier: NCT01972152).
4. Pancreatectomy
When patients are not responsive to medical treatment and cannot be weaned off treatment with intravenous glucose infusions, pancreatectomy should be considered. When a focal lesion is identified preoperatively, partial pancreatectomy is the treatment of choice. However, the lesion is not always visible or palpable at the site indicated by 18F-DOPA PET. Although intraoperative sonography can aid in identification75), repeated intraoperative biopsy may be necessary to help surgeons determine the extent of pancreatectomy. Such a treatment is made possible only by a multidisciplinary team composed of surgeons, radiologists, pediatric endocrinologists, and pathologists who are well experienced in the treatment of CHI76,77). When a focal lesion is identified in the body or tail of the pancreas using 18F-DOPA PET, resection is relatively easy; even if the exact location of the focal lesion cannot be identified, distal pancreatectomy of <70% typically cures the patient without a risk of postoperative diabetes. On the contrary, when the lesion is identified in the head of the pancreas, resection may be difficult without damaging important adjacent structures such as the main pancreatic duct or the common bile duct. In those cases, pancreatic head resection with Rouxen-Y reconstruction of distal pancreatojejunostomy has been proposed78). For patients with diazoxide-unresponsive diffuse CHI, extended resection of the pancreas is still needed. Even in these cases, near-total pancreatectomy should be avoided as much as possible in order to reduce the development of postsurgical diabetes79,80). A 70%-90% resection may be considered to reduce the pancreatic mass and to facilitate medical management.
Other unsolved questions and future perspectives
1. Causes of the remaining 50% of persistent CHI
At present, even with the most comprehensive molecular analysis, mutations in known causative genes cannot be identified in 21.3% of patients26). When confined to diazoxide-responsive cases, mutations are not identified in 53%. Therefore, if we assume that all persistent CHI is genetic in origin, there must be unidentified causative genes. In an effort to address this issue, Proverbio et al.81) analyzed 17 families with CHI who lacked mutations in ABCC8/KCNJ11 using a combination of transmission disequilibrium tests and whole-exome sequencing and reported 21 novel genes as possible candidates. Although none of these have been confirmed as causative, further efforts employing next-generation sequencing may answer these questions.
Using next-generation sequencing, Flanagan et al.82) took a different approach of sequencing the entire genomic region of the ABCC8 and HADH genes82). By this strategy, they identified deep intronic mutations of both genes causing CHI, c.1333-1013A>G in ABCC8 and c.636þ471G>T HADH. Surprisingly, these mutations were common in the Irish and Turkish populations, accounting for 14% of focal hyperinsulinism cases and 32% of subjects with HADH mutations.
2. Causes of transient CHI (a hypothesis)
Transient CHI is common in infants who were born small for their gestational ages (SGA) or in those with perinatal stress. However, little is known about its cause. SGA infants are in a hypoxemic condition in utero83). Because β-cell function is inhibited by hypoxia-inducible factor 1α (HIF1α)84,85), a sudden increase in the oxygen tension at delivery may downregulate HIF1α leading to hyperinsulinemia. In line with the observation that oxygenation of fetal blood improves with gestational ages83), it has been reported that blood insulin levels at birth correlate with the gestational age of the infants: 9.2 µIU/mL for full term; 10.3 µIU/mL for early term; 13.2 µIU/mL for late preterm; and 18.9 µIU/mL for early preterm86). Hyperinsulinemia at birth, therefore, is a common finding in newborns with a lower birth weight.
3. Mechanism of spontaneous remission of CHI
Both diffuse and focal HI resolve spontaneously over time87). A previously proposed mechanism for spontaneous remission of CHI is apoptotic death of insulin-oversecreting β-cells88). However, the initial event could be functional shutdown of insulin secretion rather than apoptotic cell death because the abnormal β-cells could still be observed by 18F-DOPA PET at an early stage of the spontaneous remission of focal KATP-CHI89). Manipulating the process of functional shutoff could be an attractive treatment option for CHI.
4. Novel medications for diazoxide-unresponsive CHI
1) Novel somatostatin analogues
Novel somtatostatin analogues have been successfully used for CHI or other forms of HI, including lanreotide90,91) or long-acting octreotide92) for CHI. In addition, pasireotide has been tested for severe postgastric bypass HI93). Although octreotide and lanreotide have affinities for somatostatin receptors SSTR2 and SSTR5, pasireotide has a broader spectrum of activity for other types of SSTRs94).
2) Small molecule corrector of KATP-channel CHI
The search for small molecules to treat CHI is fueled by previous efforts to correct the trafficking defect of the cystic fibrosis transmembrane conductance regulator, which is deficient in patients with cystic fibrosis. The idea is to use small molecules as pharmacological chaperones to correct the trafficking defect and help their expression to the cell surface95). This strategy is applicable to certain mutations of the KATP-channel genes. Thus far, sulfonylureas96) and carbamazepine97) have been successfully used to correct the trafficking defects of mutations within the transmembrane domains 0 and 1 (TMD0, TMD1) of ABCC8.
3) Glucagon-like peptide 1 (GLP1) receptor antagonist
GLP1 is secreted from the L-cells of the small intestine and binds to the GLP1 receptors in pancreatic β-cells, thereby stimulating the secretion of insulin (the incretin pathway). This pathway has a role in the amplification of postprandial insulin secretion and has been the target of novel treatments for type 2 diabetes. An antagonist of the GLP1 receptor, exendin9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39), has been shown to be effective for the treatment of CHI in an open-label, randomized clinical trial98). Although the effect was not complete, and monotherapy of CHI with this class of medication appears impractical, GLP1 receptor antagonists may have a role in adjunctive treatment of CHI.
4) Mammalian target of rapamycin (mTOR) inhibitors
mTOR is a member of the serine/threonine kinase family and is induced by amino acids (arginine and branched-chain amino acids), stress, high-energy status, oxygen, and growth factors. mTOR is complexed with regulatory-associated protein of mTOR (Raptor), mammalian LST8/G-protein β-subunit-like protein (mLST8/GβL), PRAS40, and DEPTOR to form the mTORC1 complex. Alternatively, mTOR is complexed with mLST8/Gβ, rapamycin-insensitive companion of mTOR (Rictor), and mammalian stress-activated protein kinase-interacting protein 1 (mSIN1) to form mTORC2 and is active in a variety of cellular mechanisms, including protein synthesis, cell proliferation, or cell survival. Therefore, mTOR inhibitors have been widely used to treat neoplasms. In terms of glucose metabolism, activation of mTORC1 is known to cause increased glucose uptake and glycolysis via HIF1. In addition, mTORC2 is known to play an important role in maintaining the β-cell mass through the phosphotidylinositol-3-kinase/mTORC2/AKT signaling pathway99). Sirolimus, one of the mTOR inhibitors, was successfully used to treat patients with diazoxide unresponsive CHI100). These classes of medications therefore may have a role in the treatment of CHI as well.
Acknowledgments
This work was supported by a Grant-in-aid for Scientific Research from the Ministry of Health, Labour and Welfare of Japan (Research on Measures for Intractable Diseases 2012-070).
Notes
No potential conflict of interest relevant to this article was reported.