Genetic evaluation using next-generation sequencing of children with short stature: a single tertiary-center experience
Article information

Abstract
Purpose
We used next-generation sequencing (NGS) to investigate the genetic causes of suspected genetic short stature in 37 patients, and we describe their phenotypes and various genetic spectra.
Methods
We reviewed the medical records of 50 patients who underwent genetic testing using NGS for suspected genetic short stature from June 2019 to December 2022. Patients with short stature caused by nongenetic factors or common chromosomal abnormalities were excluded. Thirty-seven patients from 35 families were enrolled in this study. We administered one of three genetic tests (2 targeted panel tests or whole exome sequencing) to patients according to their phenotypes.
Results
Clinical and molecular diagnoses were confirmed in 15 of the 37 patients, for an overall diagnostic yield of 40.5%. Fifteen pathogenic/likely pathogenic variants were identified in 13 genes (ACAN, ANKRD11, ARID1B, CEP152, COL10A1, COL1A2, EXT1, FGFR3, NIPBL, NRAS, PTPN11, SHOX, SLC16A2). The diagnostic rate was highest in patients who were small for their gestational age (7 of 11, 63.6%).
Conclusions
Genetic evaluation using NGS can be helpful in patients with suspected genetic short stature who have clinical and genetic heterogeneity. Further studies are needed to develop patient selection algorithms and panels containing growth-related genes.
Highlights
· Genetic evaluation can be helpful in patients with suspected genetic short stature. We performed next-generation sequencing (NGS) in 37 patients, and overall diagnostic yield was 40.5% (15 of 37). It is necessary to develop patient selection algorithms or guidelines for genetic testing.
Introduction
Short stature, defined as a height more than 2 standard deviations below the mean of the population adjusted for age and sex, is a common reason for referral to pediatric endocrinologists. Some children with severely short stature are vulnerable to various developmental, psychological, and social problems [1]. Growth is a complex process that is influenced by multiple factors, such as heredity, nutrition, hormones, and the environment. Nevertheless, genetic variations are important, accounting for 60%–80% of linear growth, and more than 700 genes that affect the growth process have been identified [2,3]. Genetic factors that influence height include chromosomal abnormalities, pathogenic variants of a single gene, and polygenetic predispositions [4,5]. With advances in nextgeneration sequencing (NGS) technologies, particularly targeted panel tests and whole exome sequencing (WES), genetic testing has become an important method in pediatric endocrinology clinics for identifying the causes of patients' short stature. The role of WES is expanding not only to increase the diagnostic rate but also to discover new causative genes in patients with short stature of unknown cause [6-8].
Short stature can also be caused by nongenetic causes, such as malnutrition, chronic systemic diseases, endocrine or metabolic disorders, and psychological deprivation. Some patients with short stature show other clinical abnormalities, including skeletal deformity, facial dysmorphism, developmental delay (DD), and other organ defects. In many cases, the genetic and nongenetic causes of short stature might not be clearly distinguishable. Because no clinical guidelines or diagnostic algorithms have yet been established to determine when and how to apply genetic testing to patients with short stature, the decision to perform genetic tests for such patients depends on their family history, accompanying clinical signs, and radiologic findings. The diagnostic rate of genetic testing using NGS in children with short stature has been reported to vary from 9% to 46.2%, depending on the characteristics of the patient group [6,9-13]. The choice of an appropriate genetic testing method, considering the diagnostic rate and cost, is also challenging for clinicians. To obtain a high diagnostic yield from genetic testing, it is important to select patients who will most likely benefit from it and implement a well-designed panel or WES [10,13]. In this study, we used NGS (targeted panel sequencing and WES) to investigate the genetic causes of short stature in 37 patients at a single tertiary center in Korea, and we describe their phenotypes and various genetic spectra.
Materials and methods
1. Selection and description of participants
This study was approved by the Institutional Review Board (2023-01-017-000) of Inha University Hospital. We reviewed the medical records of 50 patients who visited our pediatric endocrinology clinics from June 2019 to December 2022 and underwent genetic testing for suspected genetic short stature.
The exclusion criteria were as follows: short stature caused by (1) chromosomal aneuploidy or abnormalities (such as Down syndrome, Turner syndrome, chromosome 22q11.2 deletion); (2) multiple pituitary hormone deficiency (MPHD); (3) chronic systemic disorder such as chronic renal failure, chronic inflammatory disease, or inherited metabolic disorder; and (4) growth failure secondary to psychological factors.
We enrolled 37 patients from 35 families in this study. We obtained written informed consent from all parents or gardians before genetic testing using NGS. In all cases, conventional methods such as karyotyping and single gene sequencing failed to determine the genetic cause of short stature. All enrolled patients met one or more of the following criteria: (1) family history of short stature with similar phenotypes; (2) severe short stature, defined as a height-standard deviation score (Ht-SDS) of less than -3; (3) additional congenital anomalies or facial dysmorphisms; (4) evidence of skeletal deformities; and (5) associated intellectual disability (ID) or developmental delay (DD).
We assessed family history through three generations, including parental height for the calculation of midparental height (MPH), and evaluated the patient's detailed clinical phenotypes. If skeletal deformity was suspected, a radiological skeletal survey was performed.
Height and weight are expressed as standard deviation scores (Ht-SDS and Wt-SDS) and were calculated based on growth standards for Korean children and adolescents [14]. MPH was calculated as follows: for females, (maternal height + paternal height − 13 cm)/2, and for males, (maternal height + paternal height + 13 cm)/2. Small for gestational age (SGA) was defined as birth weight below the third percentile of a standard sexspecific birth weight curve for gestational age in South Korea [15]. Insulin-like growth factor-1 (IGF-1) SDS at the first visit/referral or before growth hormone (GH) treatment was calculated according to the Korean standard reference [16]. GH deficiency (GHD) was confirmed, via 2 different GH stimulation tests using insulin, arginine, or L-dopa, as a peak GH response of <10 ng/mL in both tests.
2. Genetic analysis
In this study, we administered 1 of 3 genetic tests (targeted panel sequencing using 1 of 2 different panels or WES) to each patient according to their phenotypes and accompanying symptoms. After obtaining written informed consent from parents or guardians, genomic DNA was extracted from the peripheral blood of the patients and, where possible, their parents and/or siblings. Among the 37 enrolled patients, 12 underwent panel sequencing using a short stature-targeted panel (129 genes, Supplementary Table 1), 7 patients with suspected skeletal deformity underwent panel sequencing using a skeletal dysplasia-targeted panel (433 genes, Supplementary Table 2), and 18 patients underwent WES.
For the targeted panel sequencing, all exons of the target genes were captured using a Celemics custom panel (Celemics, Seoul, Korea) or an Integrated DNA Technologies (IDT) custom panel (IDT, Coralville, IA, USA). Sequencing was performed on the Illumina NextSeq platform (Illumina, San Diego, CA, USA) and generated 2×150 bp paired-end reads. DNA sequence reads were aligned to the reference sequence based on the public human genome build UCSC GRCh37/hg19. Alignment was performed with BWA-MEM, and duplicate reads were marked with biobambam2. Base quality recalibration and variant calling were performed with the Genome Analysis Toolkit, and annotation was performed with Variant Effect Predictor and dbNSFP. Sequence variants were classified using the ACMG/ AMP guidelines [17].
For WES, genomic DNA was extracted from proband blood. All exon regions of all human genes (~22,000) were captured by a Twist Human Core Exome kit (Twist Bioscience, South San Francisco, CA, United States). The captured regions of the genome were sequenced using a Novaseq 6,000 sequencing machine (Illumina). The raw genome sequencing data analysis, including alignment to the GRCh37/hg19 human reference genome, variant calling, and annotation, was conducted using open-source bioinformatics tools and in house software. We extracted evidence data on the pathogenicity of variants from previous studies and disease databases, particularly ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and UniProt (https://www.uniprot.org/). For the screening and reading of genetic mutations, EVIDENCE v.3.218)), an automatic mutation analysis system developed by 3billion, was used. Sequence variants were classified using the ACMG/AMP guidelines [17,19,20].
All suspected pathogenic/likely pathogenic (P/LP) variants were confirmed by Sanger sequencing, and copy number variants were identified by chromosomal microarray (CMA). According to the inheritance patterns of the disease, results were considered positive if one or 2 P/LP variants were identified in the disease-related genes. We report cases without a variant of uncertain significance or a P/LP variant as negative results.
3. Statistical analysis
Fisher exact test and the Wilcoxon rank-sum test were performed between the groups. Results with P<0.05 were considered statistically significant. All analyses were performed using IBM SPSS Statistics ver. 26.0 (IBM Co., Armonk, NY, USA).
4. Ethical statement
This study was performed according to the Helsinki Declaration (http://www.wma.net/en/30publications/10policies/b3/) and was approved by the Institutional Review Board of Inha University Hospital (2023-01-017-000). Written informed consent was obtained from all subjects.
Results
1. Patient characteristics
We included 37 Korean patients (17 females and 20 males) from 35 nonconsanguineous families in this study. Their median age at diagnosis was 10.5 years (range, 6 months to 17.5 years), and their mean Ht-SDS at diagnosis was -3.00±0.85. Patient demographics, auxological profiles, and clinical characteristics are listed in Table 1. Severe short stature (18 of 37, 48.6%) and facial dysmorphism (17 of 37, 45.9%) were the most common clinical manifestations. Other clinical manifestations included skeletal deformities (13 of 37, 35.1%), ID/DD (13 of 37, 35.1%), SGA (11 of 37, 29.7%), family history (10 of 37, 27%), and congenital anomalies (6 of 37, 16.2%). The 37 patients with various clinical features were divided into 2 groups, one containing those with congenital anomalies, facial dysmorphism, skeletal deformities, or ID/DD (syndromic group), and one containing those with only a family history or severe short stature (nonsyndromic group). Their diagnostic rates and clinical characteristics are compared in Table 1. The basal characteristics did not differ significantly between the groups. The diagnostic rate was higher in the syndromic group than the nonsyndromic group (46.1% vs. 27.2%, P<0.01) (Table 1).

Clinical characteristics of all patients
2. Molecular diagnosis of patients
Genetic analyses using NGS and further validation studies (Sanger sequencing or CMA) identified 13 causative genes in 15 patients (7 females, 8 males). The overall diagnostic yield was 40.5%, with the highest diagnostic yield of 57.1% for targeted skeletal dysplasia panel sequencing, 41.6% for targeted short stature panel sequencing, and 27.7% for WES. The genetic variants of the 15 patients with confirmed clinical and genetic diagnoses are summarized in Table 2. The identified genes and final diagnoses of those patients were as follows: 2 Noonan syndrome, (MIM#164790 and MIM#163950) caused by an NRAS variant and PTPN11 variant, respectively; 2 hypochondroplasia (MIM#146000) by FGFR3 variants; 2 Seckel syndrome 5 (MIM#3613823) in siblings by CEP152 variants; short stature and advanced bone age, with or without early-onset osteoarthritis and/ or osteochondritis dissecans (MIM#165800) by an ACAN variant; KBG syndrome (MIM#148050) by an ANKRD11 variant; Coffin-Siris syndrome 1 (MIM#135900) by an ARID1B variant; osteogenesis imperfecta type 3 (MIM#259420) by a COL1A2 variant; metaphyseal chondrodysplasia, Schmid type (MIM#120110) by a COL10A1 variant; exostoses, multiple, type 1 (MIM#133700) by an EXT1 variant; Cornelia De Lange syndrome 1 (MIM#122470) by an NIPBL variant; Leri-Weill dyschondrosteosis (MIM# 127300) by a SHOX variant; and X-linked Allan-Herndon-Dudley syndrome (MIM#300523) by an SLC16A2 variant. One gene (CEP152) was inherited in an autosomally recessive manner, 1 gene (SLC16A2) was inherited in an X-linked manner, 1 gene (SHOX) was inherited in a pseudo-autosomally dominant manner, and the remaining 10 genes were inherited in an autosomally dominant manner.
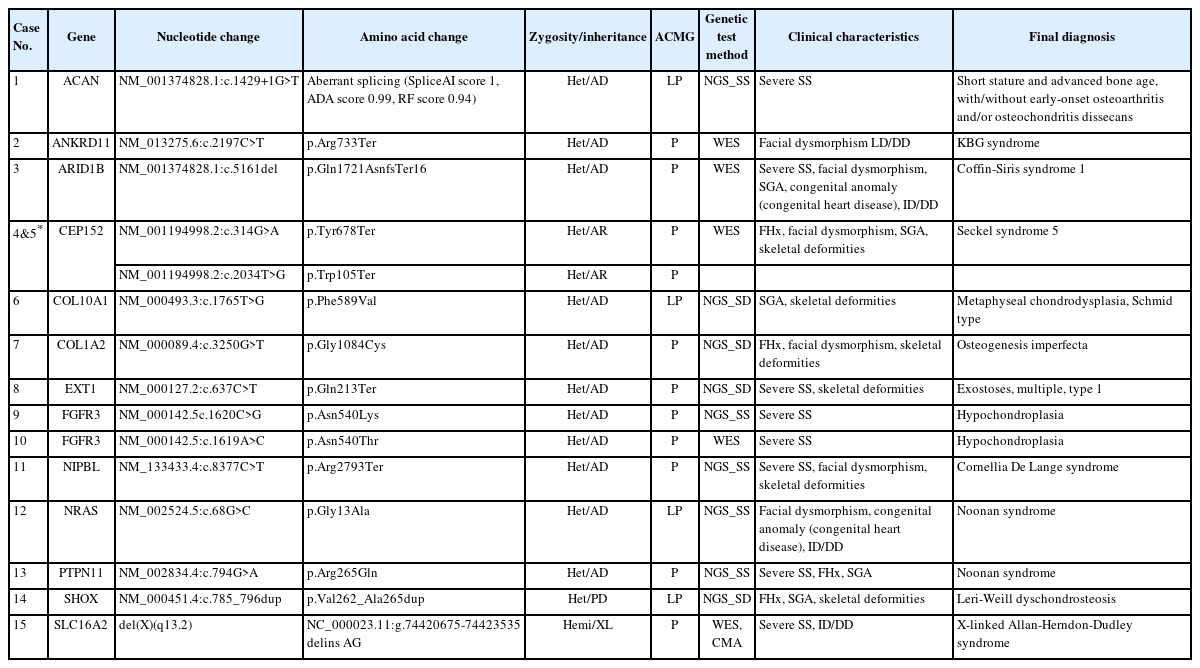
Genetic variants of 15 patients with confirmed diagnoses
Table 3 shows the clinical characteristics and phenotypes of the positive and negative groups. The basal characteristics and auxological profiles did not differ significantly between them. In the positive group, only 2 patients (13.3%) received GH stimulation tests, and neither of them was diagnosed with GHD. On the other hand, most of the negative group received GH stimulation tests, and 64.7% of them were diagnosed with GHD. Among the major clinical features, SGA (46.6% vs. 18.1%, P=0.031) and skeletal deformity (53.3% vs. 22.7%, P=0.027) occurred more frequently in the positive group. Other clinical characteristics did not differ significantly between the groups (Table 3).

Comparison of clinical characteristics between groups
Discussion
This study used NGS to explore diagnoses of genetic short stature in 37 patients with short stature and various clinical features at a single tertiary center in South Korea. In this study, we established a molecular diagnosis in 15 patients, who together had variations in 13 different genes. These results suggest that growth is affected by various genetic factors and that patients with genetic short stature form a highly heterogeneous group. This genetic heterogeneity makes it difficult to perform genetic testing in patients with short stature and is a cause of low diagnostic yield.
Several studies have used NGS to identify genetic causes in patients with short stature [6,8-13,21,22]. Because the number of causative genes is large and patient phenotypes are diverse, the diagnostic yield differed depending on the genes included in the panel and the patient group selected. Hauer et al. [8] reported an additional diagnostic yield of 16.5% when WES was performed on 200 patients (and their families) whose disease cause was not identified by systemic phenotyping. In a Chinese study, targeted panel sequencing of 166 growth-related genes was applied to 91 patients with idiopathic short stature (ISS), and the genetic diagnostic rate was low (9%) [11]. In a recently published domestic study of 114 Korean patients with ISS and isolated GHD, targeted panel sequencing of 96 genes was performed, and the diagnostic rate was 10%, similar to the previous study [9].
However, Kim et al. [13] conducted targeted exome sequencing on 15 Korean patients with suspected syndromic growth disorder and reported a diagnostic rate as high as 46.2%. Recently, Li et al. [6] published a study in which targeted NGS and WES were performed in 814 patients with short stature and other clinical symptoms, the largest cohort to date. Their overall diagnostic yield was 44.3%, with 111 causative genes in 361 patients. Among the various clinical features associated with short stature, ID/DD had the highest diagnostic rate at 70%, followed by skeletal deformity at 64.7%, microcephaly at 56.3%, congenital anomalies or dysmorphic features at 56.2%, MPHD at 36.4%, severe isolated GHD at 25%, and SGA without catch-up growth at 20.5%. Patients with severe short stature without additional phenotypes had the lowest diagnostic rate (11.2%). In this study, the overall diagnostic rate was 40.5%, which is similar to that reported in other studies that targeted patients with suspected syndromic genetic stature. Although we included a small number of patients, the diagnostic rate of SGA (7 of 11, 63.6%) was the highest in this study, followed by skeletal deformities (8 of 13, 61.5%), ID/DD (5 of 13, 38.4%), and congenital anomalies (2 of 6, 33.3%) or facial dysmorphism (5 of 17, 29.4%).
Genes affect short stature by multiple complex mechanisms that can generally be classified as follows [23,24]: (1) Defects in the GH–IGF-1 axis; (2) defects in paracrine signaling of the growth plate; (3) defects in cartilage extracellular matrix (ECM) maintenance; (4) defects in fundamental cellular (intracellular) pathways, including RASopathies, DNA repair mechanisms, and transcriptional factors; and (5) chromosomal abnormalities, copy number variations, and imprinting disorders. In this study, patients with isolated severe GHD, MPHD, and GH insensitivity (GHI) were excluded, and genes related to the GH– IGF-1 axis were not identified. Defects in fundamental cellular (intracellular) pathways were the most important genetic cause of short stature in this study. This classification contains a large number of genes, and the associated pathophysiologic mechanisms and clinical diseases are heterogeneous. In previous studies, this pathway accounted for the largest portion of genetic causes in patients with short stature [6,8,10,11]. The phenotypes of these patient groups were very diverse, but they were often syndromic short stature accompanied by clinical features other than those of ISS [10]. Among them, abnormalities in the Ras-MAPK pathway, including atypical Noonan syndrome, is an important determinant of genetic short stature, and 2 of the patients in this study belonged to that category.
Another important genetic cause of short stature is defects in cartilage ECM construction and maintenance. Five patients with that cause were included in this study, and most of them had skeletal deformity. The ACAN gene, which encodes the proteoglycan core protein aggrecan, is an important causative gene for short stature and has recently received attention. Aggrecan is mainly expressed in cartilage growth plates, and the pathogenic variant of ACAN can accelerate bone mutations and cause early growth cessation [25]. A heterozygous variant of the ACAN gene was found at a high frequency (2.5%) in both nonsyndromic and syndromic short stature [8,9,11] and was similar to the SHOX gene (2.4%), which is known to be the most common single gene defect in short stature [26]. These patients might have severe short stature with accelerated bone age, and other symptoms, such as early-onset osteoarthritis, should be noted.
Short children with SGA also form a clinically heterogeneous group, and various factors are known to be involved in the failure of catch-up growth [27]. As mentioned above, disorders of the GH–IGF axis, paracrine signaling, cartilage ECM, and fundamental cellular (intracellular) pathways can also be affected in children with SGA without catch-up growth. The highest diagnostic rate associated with SGA in this study is thought to reflect our choice to perform genetic testing on SGA patients only when they also had other clinical signs, such as skeletal deformity, facial dysplasia, and ID/DD. Therefore, genetic testing can be considered for short children with SGA accompanied by other symptoms after ruling out maternal/placental factors and common disorders related to SGA through careful examination.27,28
Genetic diagnosis is important in short stature, especially in children, because it can influence treatment decisions and improve prognosis by preventing possible accompanying symptoms. GH treatment (GHT) is the most important treatment for children with short stature. However, many cases of genetic short stature do not have GHD; in such cases, GHT is often not helpful in improving the patient's adult height. GHT can be burdensome to families because of its costs and need for daily injections for children. GHT might not be an appropriate treatment option for patients with GHI or skeletal deformities such as severe scoliosis. In our cohort, 51.4% of the patients were tested with GH stimulation, and the prevalence of GHD was as low as 29.7%. Only 13.3% of the positive group was tested with GH stimulation, and no patient was diagnosed with GHD. On the other hand, in the negative group, most of the patients received the GH stimulation test, and 64.7% of them were diagnosed with GHD and treated with GH. These findings suggest that appropriate genetic evaluations can help to determine whether GH stimulation tests or GHT should be performed. In addition, some of the patients with definite molecular diagnoses received multidisciplinary care and additional tests for possible accompanying symptoms, including developmental disorders, other endocrine disorders, and skeletal abnormalities. Accordingly, prognosis and followup management were planned, and genetic counseling was performed.
When applying a genetic test using NGS, the decision to perform a targeted panel test or WES is important for maximizing the diagnostic rate. In this study, 2 panels were used based on the accompanying clinical features of the patient, and WES was performed when a targeted panel could not be specified due to the variety of clinical features. In previous studies, the diagnostic rates of panel tests were similar to or slightly higher than those of WES [6,8,12]; however, in our study, the diagnostic rate of the panel tests was considerably higher. This finding indicates that performing genetic evaluations according to patient phenotype is an important way to optimize the diagnostic rate.
For patients with multiple clinical features, WES can be a suitable alternative. However, WES is not available except for research purposes in Korea, so it is important to develop panels that include a wide range of genes that can accompany short stature. Furthermore, genes related to growth are still being discovered; therefore, panel designs will need to be updated regularly to increase the diagnostic rate.
Our study has some limitations. First, this study was conducted in a single center for a relatively short period of time, and the number of enrolled patients was not sufficient to demonstrate the various genetic spectra of short stature. In addition, because this study was conducted retrospectively, the clinical characteristics of the patients were heterogeneous, and selection bias was a possibility. Finally, the non-uniformity of NGS testing methods used might have affected the diagnostic rate.
In conclusion, we performed genetic evaluations using NGS in 37 patients with suspected genetic short stature. We established a molecular diagnosis in 15 (P/LP variants in 13 causative genes) of those 37 patients, for an overall diagnostic yield of 40.5%. It is necessary to develop patient selection algorithms or guidelines for genetic testing, develop a panel that includes growth-related genes with high diagnostic rates, and conduct large-scale studies to understand the characteristics of patients with genetic short stature.
Supplementary Material
Supplementary Tables 1 and 2 can be found via https://doi.org/10.6065/apem.2346036.018.
Lists of including genes in a short stature targeted panel
Lists of including genes in a skeletal dysplasia targeted panel
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This work was supported by Inha University/Inha University Hospital Research Grant.
Data availability
The data that support the findings of this study can be provided by the corresponding author upon reasonable request.
Author contribution
Conceptualization: SJK; Data curation: SJK, EJ; Formal analysis: SJK; Funding acquisition: SJK; Methodology: SJK, CAS; Project administration: SJK, JEL; Visualization: SJK; Writing - original draft: SJK; Writing - review & editing: SJK, JEL
Acknowledgements
The authors thank all the patients and their caregivers for their involvement in this study.