Short stature with low serum alkaline phosphatase activity: a case report of hypophosphatasia
Article information

Abstract
Hypophosphatasia (HPP) is a rare condition characterized by abnormal bone mineralization. The manifestations of HPP vary from no symptoms to intrauterine fetal death; short stature is another indication of HPP. A 3 ½-year-old boy presented with short stature, transient hypercalcemia, and mild gait disturbance without definite bony deformity. Laboratory examination revealed transient hypercalcemia, normal phosphorous and 25-hydroxy vitamin D levels, and mildly low alkaline phosphatase levels. A targeted next-generation sequencing panel associated with inborn errors of metabolism revealed a pathogenic heterozygous mutation in the ALPL gene, c.979T>C (p.Phe327Leu). When a child visits a hospital with short stature, decreased height velocity, and low alkaline phosphatase level, clinicians should consider the possibility of HPP even if definite skeletal dysplasia is not evident.
Highlights
· Hypophosphatasia (HPP) is a rare genetic disorder characterized by abnormal bone mineralization, which caused by loss-of-function mutations in the ALPL gene. We report a case of HPP confirmed by genetic testing in a child who visited hospital due to short stature without skeletal dysplasia.
Introduction
Hypophosphatasia (HPP; OMIM number 146300, 241500, 241510) is a rare genetic disorder inherited in an autosomal dominant or recessive pattern, characterized by defective bone and dental mineralization. HPP is caused by loss-of-function mutations in the ALPL gene located on chromosome 1p36.1-p34, which encodes tissue-nonspecific alkaline phosphatase (TNSALP) [1]. TNSALP, a human alkaline phosphatase (ALP), is highly expressed in the liver, bone, and kidney and hydrolyzes organic phosphates, such as inorganic pyrophosphate (PPi), phosphoethanolamine (PEA), and pyridoxal-5'-phosphate (PLP), which is a major circulating form of vitamin B6. In the growth plate, TNSALP hydrolyzes PPi to inorganic phosphate, which regulates proliferation, differentiation, and mineralization of endochondral ossification [2]. Reduced TNSALP activity results in the accumulation of these substrates, leading to increased serum and urine levels. Extracellular deposition of PPi directly inhibits mineral deposition by the osteoblasts and chondrocytes (especially crystals of basic calcium phosphate hydroxyapatite) in the pericellular matrix of bone, which is associated with decreased bone matrix and bone strength, with can present as short stature [3].
HPP is associated with a wide spectrum of clinical manifestations including short stature [4,5]. In a study including Japanese patients with HPP, 21 of 52 patients showed short stature [6]. In another study, 13.6% of patients with HPP showed short stature, and the childhood form was the most prevalent at 30% [7]. Causes of short stature are numerous, such as endocrine disorders, bone diseases, and genetic disorders. However, HPP is not usually considered a differential diagnosis of short stature due to its low prevalence and vague clinical features. Recently, interest in HPP has gradually increased due to the use of genetic testing and introduction of therapeutic agents. Herein, we report a case of HPP confirmed based on genetic testing in a pediatric patient who visited our hospital for evaluation of short stature without skeletal dysplasia, which is the first case reported in Korea.
Case report
A 3 ½-year-old boy with short stature was referred to our clinic. He was born at a gestational age of 32 weeks 5 days and had a twin sister. The patient's birth weight, length, and head circumference were 1.67 kg (-0.73 standard deviation score [SDS]), 41 cm (-0.79 SDS), and 29.5 cm (-0.35 SDS), respectively (Table 1). Physical examination after birth revealed no facial dysmorphism or other accompanying malformation except hypospadias. In addition, specific skeletal deformities, such as micromelia, bowed legs, small thoracic circumference, or costochondral junctions, were not observed. He was admitted to the neonatal intensive care unit due to premature birth and development of transient tachypnea of the newborn and was treated with continuous positive airway pressure for 4 days. Chest radiography revealed no findings suggestive of thoracic and lung hypoplasia, short and thin ribs, small thorax, or lung hypoplasia. The blood tests performed on the first day after birth revealed calcium, 9.9 mg/dL (reference, 8.5−11.0 mg/dL); phosphorus, 8.1 mg/dL (reference, 5.6−10.5 mg/dL); and ALP, 187 IU/L (reference, 90−273 IU/L; Table 2). The automated auditory brainstem response and echocardiography findings were also normal.

Growth measurement of the patient

Laboratory findings of the patient
Both parents were healthy with an unremarkable medical history, and significant findings were not noted on the prenatal questionnaire. The mother was 35 years of age and had become pregnant through an in vitro fertilization-embryo transfer process. The laboratory and radiological findings were normal. The heights of the father and mother were 174 cm and 163 cm, respectively, with an estimated mid-parental height, 175 cm. The patient's twin sister had a normal birth length of 40 cm (-0.85 SDS) and a birth weight of 1.64 kg (-0.50 SDS) without any particular deformities.
The child was admitted at 4 months of age with viral enteritis and at 9 months of age for surgical correction of hypospadias. His height and weight at 4 and 9 months of age were 56 cm (-1.21 SDS) and 5.9 kg (0.47 SDS) and 69 cm (-0.07 SDS) and 8.02 kg (-0.31 SDS), respectively (Table 1). At 4 and 9 months, mild hypercalcemia was present (11.0 mg/dL and 11.2 mg/dL; reference, 8.5–11.0 mg/dL), the phosphorous levels were 6.3 mg/dL and 6.1 mg/dL (reference, 4.8–8.4 mg/dL), and the ALP levels were 157 and 153 IU/L (reference, 134–518 IU/L), respectively (Table 2) [8]. At the 20-month infant screening, his height and weight were 80.5 cm (-0.65 SDS) and 10 kg (-0.80 SDS), respectively (Table 1). At 26 months of age, he underwent rehabilitation for mild gait disturbance, tip-toeing gait, and delayed language development. Abnormal findings were not observed on several skeletal radiographs performed at our hospital.
At 3 ½ years of age, his height, weight, and body mass index were 91.3 cm (-2.28 SDS), 12.7 kg (-2.16 SDS), and 15.23 (-0.62 SDS), respectively, indicating short stature (Table 1). In contrast, his twin sister showed normal height and growth. His eating habits were good at that time, and he was active without any early loss of deciduous teeth. Laboratory test results of the patient were as follows: random growth hormone, 3.1 ng/mL (reference, 0.7−6 ng/mL); insulin-like growth factor-1 (IGF-1), 165.72 ng/mL (reference, 32.2−255.4 ng/mL); insulinlike growth factor binding protein-3 (IGFBP-3), 2.501 µg/mL (reference, 1.18−2.72 µg/mL); thyroid-stimulating hormone, 4.34 µU/mL (reference, 0.5–4.8 µU/mL); free thyroxine, 1.4 ng/dL (reference, 0.8–2.2 ng/dL); 25-hydroxy vitamin D, 118.73 nmol/L (reference, 50–250 nmol/L); calcium, 10.1 mg/dL (reference, 9.2−10.5 mg/dL); phosphorous, 4.7 mg/dL (reference, 4.3−6.8 mg/dL); parathyroid hormone, 40.0 pg/mL (reference, 11−59 pg/mL); and ALP, 125 IU/L (reference, 156−369 IU/L; Table 2). Urine PEA level was 127.6 nmol/mg creatinine (reference, 18–150 nmol/mg creatinine). Other laboratory tests, including complete blood cell count (CBC), electrolytes, protein, and albumin, were normal. His bone age was 3.2 years and 3 years according to the Tanner-White 3 method and Greulich-Pyle method, respectively.
Although random growth hormone and IGF-1 levels were within normal limits, growth hormone stimulation tests were performed due to delayed bone age and decreased height velocity. The peak growth hormone level was 6.84 ng/mL and 4.85 ng/mL in 2 provocation tests conducted using arginine and insulin, respectively, indicating growth hormone deficiency (GHD). Screening tests for inborn errors of metabolism were performed to evaluate the causes of abnormal gait and developmental delay, revealing a decreased level of serum acetyl-carnitine at 1.86 nmol/mL (reference, 2.00–27.57 nmol/mL) and an increased level of urine glutaric acid at 10.8 mmol/mol creatinine (reference, <5.3 mmol/mol creatinine). Sellar magnetic resonance imaging (MRI) showed no specific findings in the pituitary gland.
Due to the short stature, slow growth velocity, low ALP levels, abnormal gait, and increased levels of 2 metabolites, a targeted next-generation sequencing panel associated with inborn errors of metabolism genes including ACADM, ACADVL, ACAT1, ALPL, ASL, ASS1, BCKDHA, BCKDHB, CBS, DBT, FAH, GCDH, HADHA, HADHB, HLCS, HMGCL, IVD, MMAA, MMAB, MMUT, PAH, PCCA, PCCB, and SLC22A5 was conducted after obtaining informed consent from the parents. Genomic DNA was obtained from peripheral blood and analyzed using sequencing with synthesis technology. The size of the capture target region was 639 genes (1,979,160 bp) with a 714.40-fold mean sequencing depth. The panel test identified a heterozygous mutation c.979T>C (p.Phe327Leu) in ALPL, which was classified as a pathogenic variant according to the American Medical College of Medical Genetics and Genomics guidelines (Fig. 1). Genetic testing of the parents was not performed.

A heterozygous mutation in the ALPL gene, c.979T>C (p.Phe327Leu).
Treatment was started with growth hormone injection at 0.33 mg/kg/day. After 3 months, the patient's height and weight were 96.2 cm (-1.74 SDS) and 13.8 kg (-1.90 SDS), respectively, and adverse effects did not occur. Recently, the patient visited the ER complaining of pain in his right arm after hitting a door. Radiographs showed a subtle supracondylar fracture of the right humerus (Fig. 2). However, obvious osteoporosis or bone deformity such as radiolucency of the tongue, metaphyseal fraying, or apparent physeal widening was not observed. Because the patient was diagnosed with HPP, regular follow-ups and a bone mineral density test were subsequently conducted.
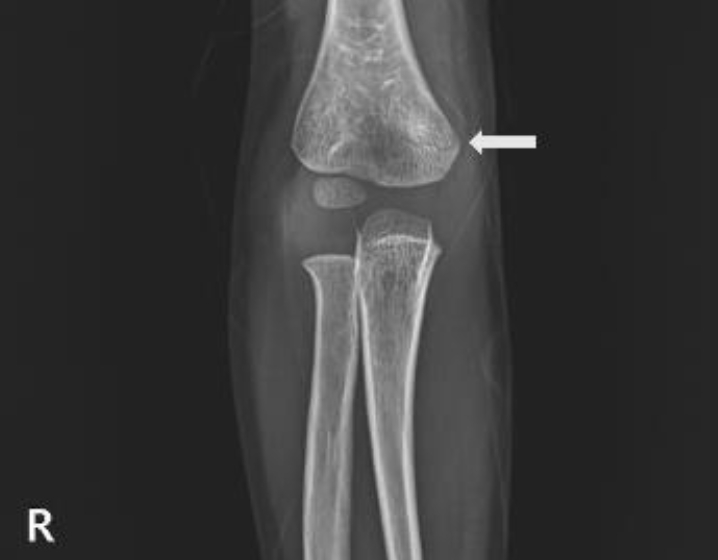
Humerus radiographs showing a right humerus supracondylar fracture at 3 years 11 months of age (white arrow).
The Institutional Review Board (IRB) of Keimyung University Dongsan Hospital (IRB No. 2022-12-034) approved this retrospective study that was conducted in accordance with the World Medical Association Declaration of Helsinki. Informed consent was obtained from the patients' parents.
Discussion
HPP has a wide spectrum of clinical manifestations, and prognosis is based on the ALP level and age of onset [9]. Accordingly, HPP is classified into 6 phenotypes: lethal perinatal, benign perinatal, infantile, childhood, adult, and odontohypophosphatasia. In the childhood form diagnosed in our patient, clinical symptoms usually appear 6 months after birth. Signs and symptoms include a delay in walking, short stature, and gait disturbance; spontaneous remission is possible [5].
Although the exact mechanism is unclear, short stature is frequently observed in patients with HPP. In a previous study, short stature was observed in 21 of 52 patients with HPP (perinatal lethal form, 5; benign perinatal form, 8; infantile form, 2; childhood form, 6). The prevalence of short stature was highest in the childhood form (66%, 6 of 9), and approximately half of the patients with a short stature were diagnosed with GHD [6]. Therefore, in addition to GHD, HPP may contribute to short stature. For instance, inhibition of chondrocyte ossification due to accumulation of PPi caused by low activity of TNSALP may be associated with short stature.
Our patient had a normal weight and height from birth to 20 months; however, at 3 ½ years of age, decreased growth rate was observed, with both weight and height less than -1.88 SDS. Although our patient carried a heterozygous mutation in the ALPL gene and accompanying GHD, he was very active and showed no findings of central obesity. He showed normal basal IGF-1 and IGFBP3 levels as well as normal MRI findings. The patient also developed mild hypercalcemia at 4 months of age, abnormal gait at 26 months of age, short stature and decreased height velocity at 3 ½ years of age, distal humerus supracondylar fracture due to minor trauma at 3 years 11 months of age, and persistently low ALP levels, consistent findings of HPP with short stature.
The primary clinical impression of HPP is based on blood tests, clinical symptoms, and imaging and is confirmed using genetic testing. Measurement of ALP level is important in HPP but may be affected by age, sex, drugs, transfusion, and thyroid hormones; thus, HPP must be thoroughly evaluated using multiple criteria. ALP levels are physiologically higher during periods of life when bone metabolism is active, such as growth spurts. Several clinical conditions are associated with low ALP levels, including massive transfusion; disorders such as celiac disease, severe anemia, malnutrition, and hypothyroidism; and drugs (bisphosphonates, glucocorticoids, estrogens, and omeprazole). Several additional blood tests, such as CBC, liver function test, thyroid hormone test, and nutrition blood test, can help differentiate between these diseases. In our patient, there was no evidence of these diseases. In addition, the reference range of ALP varies from study to study, and appropriate reference values remain to be chosen. Colantonio et al. [8] reported a normal range of ALP based on age; in our patient, the normal range at 3 ½ years of age was 156–369 IU/L. However, reference ranges of 153–410 IU/L and 135–320 IU/L at 3 ½ years of age have been suggested in other studies (Table 3) [10,11]. In HPP, ALP levels may be inversely correlated with the severity of the condition. In a Japanese study, patients with relatively high ALP levels had mild symptoms and high survival rates [12]. In our patient, the ALP level was 125 IU/L (reference, 156–369 IU/L), which is close to the normal value, and he had a good prognosis. In addition, hypercalcemia and hypercalciuria may manifest due to impaired mineralization and bone resorption with increases in PLP and urinary PEA levels [13].

Reference intervals for ALP levels (IU/L) in healthy children aged 0–13 years
Based on molecular diagnosis, prevalence of mild HPP and severe HPP in Europe is 1 in 6,370 and 1 in 300,000, respectively [9]. Patients with the infantile form show no significant findings at birth; however, the clinical signs of HPP, such as skeletal system abnormalities including craniosynostosis, rickets-like deformities, calcification of cancellous bone, hypercalcemia, and respiratory depression, cause approximately 50% mortality before 1 year of age [14]. Patients with lethal perinatal or infantile HPP may develop pyridoxine-dependent epilepsy. TNSALP mediates the dephosphorylation of PLP to pyridoxal, which is then converted to PLP after crossing the blood brain barrier. PLP is a substrate involved in neurotransmitter biosynthesis. Because patients with HPP exhibit decreased TNSALP enzyme activity, PLP deficiency in the central nervous system and decreased neurotransmitter levels can result in pyridoxine-dependent epilepsy [13].
Imaging tests, such as ultrasound or radiography, may help diagnose HPP. Furthermore, HPP should be considered as a differential diagnosis when malformations are present, including rickets-like deformities such as long bone deformity (micromelia, bowed legs, diaphyseal spurs, metaphyseal lucencies, and absent ossification of whole bones), osteochondral spurs, and rachitic rosary or other problems in the skeletal system such as cranial deformity (wide sutures and fontanelles, absence of ossification of the skull base, and caput membranaceum), scoliosis, and recurrent fractures [15]. In our patient, definite skeletal abnormalities on radiographs were not observed from birth to the time he developed a humeral fracture. However, even if definite abnormal findings are not found on radiographs, the possibility of fractures should be considered compared with normal populations because the TNSALP activity is below the normal range.
If HPP is suspected, gene sequencing of the ALPL gene can be used for confirmation. The p.Phe327Leu (c.979T>C) mutation found in our patient is more frequently observed in Japan than in other countries [16]. Compound heterozygous mutations, including p.Phe327Leu (c.979T>C) and another mutation, were found in approximately 90.9% of patients with the benign perinatal form but have not been identified in perinatal severe, infantile, childhood, or adult-onset HPP [16]. In another study, the heterozygous p.Phe327Leu (c.979T>C) mutation was reportedly associated with childhood HPP and had relatively benign clinical features; the enzymatic activity of TNSALP is approximately 72%, which is slightly higher than that of other phenotypes [17]. Due to the comparatively high TNSALP activity, heterozygous p.Phe327Leu (c.979T>C) is mostly diagnosed as a compound type but was diagnosed as a missense mutation in one study [7].
Supportive treatments for HPP include mechanical ventilation for respiratory failure, administration of pyridoxine for epilepsy, and analgesics for skeletal pain. One case required bone marrow transplantation, but the outcome was not promising [18].
Since 2008, many clinical trials have been conducted on asfotase alfa (STRENSIQ), a recombinant TNSALP; the drug was approved by the U.S. Food and Drug Administration in 2015. In a study published in 2016, significant clinical benefits in skeletal development, muscle strength, respiratory function, and growth were observed after using STRENSIQ. In that study, the 1-year survival rate of patients treated with STRENSIQ for 48 weeks was 95%, which was significantly higher than that of historical controls (42%) [19]. When patients with HPP were treated with STRENSIQ, improvements in linear growth and weight gain were observed. In addition, significant increases in weight and height z-scores were observed after 6 weeks and 1.5 years of treatment, respectively. Furthermore, gross motor delays and functional disabilities, which can affect growth, improved after treatment [19]. In Korea, reimbursements have been performed after the pre-New Drug Application approval in 2020. Currently, our patient is not using STRENSIQ because it does not correspond to the indication. We will continuous to perform followed up while using growth hormone.
In conclusion, in children who visit the hospital for short stature with decreased height velocity, low ALP levels, and other symptoms such as fracture history and gait disturbance, the possibility of HPP may be considered even if definite skeletal dysplasia is not observed. To avoid a missed diagnosis, a thorough assessment of medical history, radiological examinations, and measurement of ALP levels should be included in the evaluation, and if necessary, a genetic test can be performed.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author contribution
Conceptualization: HSK, SK; Data curation: DL; Formal analysis: DL; Methodology: SYP; Visualization: DL, SYP; Writing - original draft: DL; Writing - review & editing: HSK, SK