Case report of familial hypobetalipoproteinemia: a novel APOB mutation and literature review
Article information

Highlights
· Familial hypobetalipoproteinemia is an autosomal dominantly inherited disorder of lipid metabolism characterized by a <5th percentile plasma levels of low-density lipoprotein cholesterol or total apolipoprotein B, caused by mutations in the APOB gene.
· Patients with heterozygous APOB-related familial hypobetalipoproteinemia are often diagnosed incidentally because they are usually asymptomatic or manifest with mild liver dysfunction. These patients may need genetic testing or follow-up tests on lipid profile and liver function.
To the editor,
Familial hypobetalipoproteinemia (FHBL) is an autosomal codominant inherited disorder of lipid metabolism characterized by a plasma level of apolipoprotein B (ApoB) below the 5th percentile. Low-density lipoprotein (LDL) cholesterol levels are usually between 20–50 mg/dL. The known causative genes for FHBL that have been discovered to date include APOB and PCSK9 [1,2]. In this report, we present a case of a novel mutation in the APOB gene identified in a Korean boy with low LDL cholesterol level and nonalcoholic fatty liver disease (NAFLD).
A 14-year-old Korean boy was referred to our clinic due to incidentally identified low LDL cholesterol and elevated aminotransferase. He complained of no specific symptoms or signs associated with hypocholesterolemia, including growth retardation, steatorrhea, or neurologic dysfunction. When he presented to the clinic, his height, weight, body mass index, and waist circumference were 164 cm (-0.14 standard deviation score [SDS]), 56.9 kg (0.09 SDS), 21.15 kg/m2 (0.13 SDS), and 70 cm (-0.27 SDS), respectively. No hepatomegaly was observed.
Initial laboratory tests showed low LDL cholesterol and ApoB and elevated aminotransferase levels (Table 1). Abdominal computed tomography revealed severe fatty liver and hepatosplenomegaly (Fig. 1). Tests for exclusion of other causes of fatty liver were conducted. Tests for hepatitis A, B, and C were negative, and serum antibodies related to autoimmune hepatitis were negative. Levels of alpha-1-antitrypsin and ceruloplasmin were within the normal ranges.

Patient laboratory findings and growth measurements
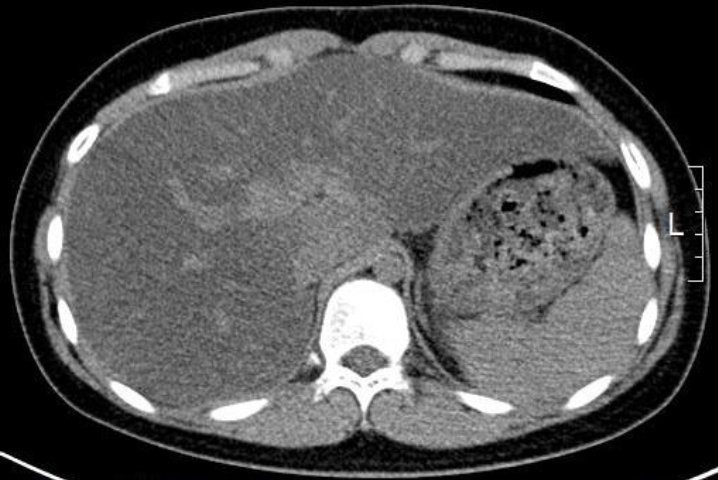
Patient abdominal computed tomographic findings. An unenhanced transverse computed tomography (CT) scan showed lower attenuation of the liver parenchyma than that of the spleen parenchyma. This finding is consistent with fatty liver. Hepatomegaly was also found in the CT scan.
Oral vitamin D supplement was started (25,000 IU/mo) for vitamin D insufficiency. He performed aerobic exercise and maintained a diet (high in fruits, vegetables, protein, and whole grains and low in saturated fats, simple sugars, and salt) for treatment of NAFLD.
Five months later, his height, weight, and body mass index were 167 cm (-0.01 SDS), 57.4 kg (-0.05 SDS), and 20.58 kg/m2 (-0.13 SDS), respectively. Laboratory tests (Table 1) revealed continued low LDL cholesterol and ApoB levels, but his aminotransferase level was within the normal range.
A multigene panel test associated with hereditary dyslipidemia was conducted. A novel heterozygous mutation for p.Lys3846Ter (c.11536A>T) was identified in the APOB gene (http://www.hgmd.cf.ac.uk, access date: July 20th, 2022). The variant was absent in control subjects according to gnomAD and the Korean Reference Genome Database. The same mutation was not detected in his parents, who both showed normal ApoB levels. The variant was classified as likely pathogenic according to American College of Medical Genetics guidelines.
After 14 months, his laboratory tests still showed low LDL cholesterol and ApoB levels (Table 1), though he again complained of no specific symptoms. His aminotransferase level was within the normal range. His vitamin E profile showed low alpha-tocopherol (4.0 mg/L; reference value, 5.5–17.0 mg/dL), for which he was started on oral vitamin E (alpha-tocopherol) 1,000 IU daily and will continue to receive regular follow-up at the clinic.
FHBL can be suspected when the total cholesterol, LDL cholesterol, and ApoB levels are below the 5th percentile for age and sex. Other supportive clinical and laboratory findings include acanthocytosis, elevated liver transaminase, failure to thrive, steatorrhea, atypical pigmentation of the retina, hepatomegaly, and fatty liver [3].
ApoB exists as two isoforms in plasma, ApoB-100 and ApoB-48 [4]. ApoB-100 is synthesized in the liver and is a major structural component of very-low density lipoprotein, intermediate-density lipoprotein, and LDL. ApoB-48 is synthesized in the intestine and is the major structural protein in chylomicrons and chylomicron remnants [5]. APOB mutation induces the production of truncated ApoB-100, which leads to low plasma concentrations of total cholesterol, triglycerides, and LDL cholesterol because of the impaired export of triglycerides by very-low density lipoprotein from the liver [6]. Accumulation of triglycerides in hepatocytes leads to NAFLD in patients with APOB-related FHBL (APOB-FHBL).
Patients with homozygous APOB-FHBL typically have symptoms of fat malabsorption, steatorrhea, diarrhea, failure to thrive, deficiencies in fat-soluble vitamins, and neurologic dysfunction. They usually present with hepatomegaly and hepatic steatosis. These patients may require a low-fat diet and supplementation with fat-soluble vitamins [3]. Patients with heterozygous APOB-FHBL are usually asymptomatic with mild liver dysfunction and hepatic steatosis. They are often diagnosed incidentally due to low cholesterol levels and generally do not require special treatment or restriction of fat intake. However, in rare cases, relatively severe nonalcoholic fatty liver develops and progresses to cirrhosis or even hepatocellular carcinoma, particularly in the presence of known risk factors such as alcohol consumption, excessive caloric intake, and liver injury. Therefore, it is important to reduce these risk factors and perform regular follow-up [3,7,8]. Recommended surveillance requires laboratory investigations including lipid profile and liver function tests every 1–2 years, and radiologic examination including hepatic ultrasonography every 3 years after 10 years of age.
In conclusion, we report a case of a boy with a novel APOB mutation. Known risk factors associated with NAFLD progression, such as obesity and insulin resistance, should be closely observed and managed, and further genetic testing may be required.
Notes
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics statement
This retrospective analysis was approved by the Institutional Review Board (IRB) of Keimyung University Dongsan Hospital (IRB No. 2022-07-047). We obtained informed consent from the patient's parents.